Keywords
INTRODUCTION
Myxoma is the most frequent primary cardiac tumor and has been diagnosed in patients over a wide range of ages, from neonates through to individuals aged 95 years. Its clinical manifestations include embolic events and obstruction of the left atrial outflow tract (dyspnea, orthopnea), as well as more general symptoms such as fatigue, myalgia, fever, and weight loss.1,2 Patients, and members of their immediate family, have been identified with recurrent myxomas which are accompanied by changes in skin pigmentation and endocrine hyperactivity, abnormalities referred to as the Carney complex.3
From January 1986 to November 2005, we studied 63 patients with an echocardiographic, surgical, and histopathologic diagnosis of cardiac myxoma. Tumor recurrence was documented in 5 (7.9%) patients. Genetic studies were performed in 3 of those patients, as well as in 11 of the remaining 58 patients. The purpose of this study is to describe the clinical, echocardiographic, and genetic characteristics of patients with recurrent myxomas and to compare them with 11 patients with isolated myxomas (Table).

METHODS
DNA was extracted from a 10 mL blood sample using conventional techniques.4 The 12 exons of the PRKAR1A gene on chromosome 17 were analyzed for polymerase chain reaction single-strand conformation polymorphism (PCR-SSCP) to detect regions with possible mutations. These regions were later sequenced using an automatic capillary sequencer. The sequences were compared with the consensus sequence and with sequences with mutations reported in the literature. When a mutation appeared, a codon by codon analysis was performed to establish the type of mutation as well as its position on the gene.5,6
The criteria proposed by Stratakis were used to establish a diagnosis of Carney's complex.7 The first consists of identifying 2 of the following alterations: skin pigmentation abnormalities, multiple tumors, and pigmented nodular adrenocortical disease. The second criterion is having one of the abnormalities mentioned concurrently with presence of the condition in a member of the patient's immediate family, or a mutation in the PRKAR1A gene.
Case 1
A 29-year-old woman with palpitations and dyspnea. The echocardiogram showed 2 tumors in the right ventricle (Figure 1), which were surgically resected. The histopathologic study confirmed the diagnosis of myxomas. Nine months later, a routine echocardiogram revealed a tumoral mass at the same site. The genetic study was normal.

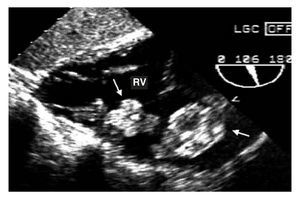
Figure 1. Transgastric echocardiogram. Two myxomas (arrows) can be observed in the right ventricle (RV).
Case 2
An 18-year-old female with a history of left amaurosis and right hemiplegia 3 years earlier. The physical examination revealed a heart murmur and a CT scan indicated multiple cerebral infarcts. The transthoracic echocardiogram showed a myxoma in the upper portion of the right atrium and another in the left atrium attached to the interatrial septum. Three myxomas were detected 3 years later: one was pediculate and located close to the left atrial appendage, the second was located in the left ventricular outflow tract, and the third was located in the right ventricular trabecular septum. The patient died after their removal due to postoperative multiple organ failure. The histopathologic study of the tumors confirmed the diagnosis of myxomas. As it was not possible to conduct a genetic study of the patient and other family members, a diagnosis of Carney complex could not be confirmed.
Case 3
A 42-year-old man with 4 children, 2 of them with lentigines on the lips. At the age of 28, the patient was diagnosed with Cushing's syndrome and lentigines of the face and hands. A few months later, a bilateral adrenalectomy was performed and the patient was diagnosed with pigmented nodular adrenocortical disease.
The following year, an echocardiogram revealed a tumor of 3.5´2.5 cm in the posterior basal region of the left ventricle. A surgical resection was performed and a histologic study indicated cardiac myxoma. After 3 years, a routine echocardiogram revealed a new myxoma in the left ventricle, though at a different site to the first. The tumor was removed but it was not possible to carry out a genetic study of the patient and other family members. Nevertheless, criteria for a diagnosis of Carney complex were met.
Case 4
A 39-year-old man with a history of right facial-corporal hemiparesis at the ages of 12 and 18 years, blood clots in the right leg, and inferior myocardial infarction. The echocardiogram revealed a 5´3 cm left atrial myxoma with 3 insertion sites including the interatrial septum. Fourteen years after tumor resection, multiple tumoral masses were found in the left atrium, some of which were attached to the interatrial septum. The others were found at different sites. The myxomas were resected and 7 years later there was no evidence of recurrence. The genetic study was normal in 3 brothers, but a mutation corresponding to a deletion of a base in the intron 2 gene PRKAR1A was identified in the patient and the patient's mother. Studies are currently underway to establish the mutation's function. Based on the above criteria, the patient was diagnosed with Carney complex.
Case 5
A 36-year-old woman with a history of heart murmur from the age of 23 years, and pigmented skin lesions. The echocardiogram showed myxoid-like tumors in the four cardiac cavities (Figure 2).8 The complete resection of the myxomas was corroborated by transesophageal echocardiography. Echocardiograms performed on the patient's parents and 5 siblings showed no cardiac masses. Two years later, further surgery was required due to the appearance of 3 new myxomas in both atria and the right ventricle. Eight years after the second surgery, surgical resection of recurring tumoral masses in the left atrium and its appendages and in both ventricles was performed. Seven years after the last surgery, a small tumor was identified in the right ventricle outflow tract (Figure 3). Findings from a physical examination and echocardiogram performed on the patient's parents and 3 of her siblings were normal. The patient's 3 children were also examined. In the youngest child, aged 6 years, no abnormalities associated with the Carney syndrome were detected. In the second child, age 8, the echocardiogram was normal, though a small tumor was detected in the scrotum, corresponding to a myxoid angiofibroma. In the third child, age 12 years, the echocardiogram showed a left atrial myxoma of 39´30 mm attached to the interatrial septum. The myoxoma was resected (Figure 4). The study showed no genetic alterations in the parents, 3 siblings, and 1 child. Both the patient and 2 of her children showed an alteration in the PRKAR1A gene, consisting of a base deletion at position 552 (552del g), corresponding to codon 155 in exon 4B. The deletion modified the reading frame and generated a stop codon and thus an incomplete, non-functional protein. This confirmed the diagnosis of Carney complex.

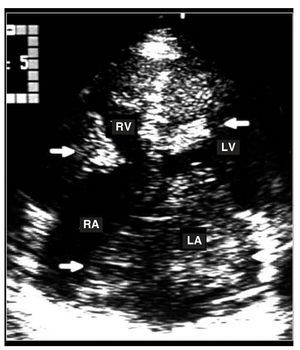
Figure 2. Apical transthoracic echocardiograpm showing several myoxomas in the 4 cardiac cavities (arrows). LA indicates left atrium; LV, left ventricle; RA, right atrium; RV, right ventricle.

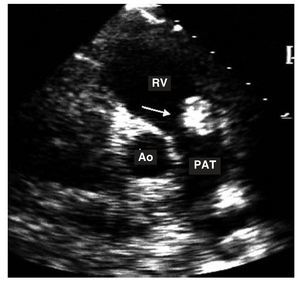
Figure 3. Transthoracic study in the transverse plane. The arrow shows a myxoma in the outflow tract of right ventricle.

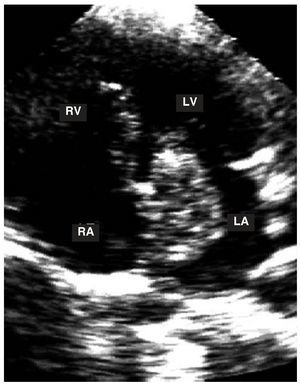
Figure 4. Apical study of the 4 chambers. A myxoma can be observed in the left atrium with a stalk attached to the interatrial septum.
RESULTS
The group with cardiac myxoma consisted of 9 women and 2 men between 25 and 56 years old. Mean age was 48 years. The myxoma was situated in the left atrium in 9 patients left and in the right atrium in 2 patients. Two of the 11 patients had a history of cerebral embolism. Follow-up after resection ranged from 1 to 17 (average, 6.3) years, and echocardiographic records showed no recurrence of tumors. A genetic study conducted in the 11 patients showed no abnormalities (Table).
DISCUSSION
In 58 of the 63 patients studied, the tumor was isolated and located in the left atrium (50 cases), the right atrium (5 cases), or the ventricles (3 cases). In 11 cases, it was possible to conduct a genetic study, but no abnormalities were found. There were no signs suggestive of Carney complex and clinical behavior was typical of an isolated myxoma.
Analysis of the 5 patients with myxomas allows certain conclusions to be drawn. In case 1, despite the unusual nature of the tumor site, the histopathologic study indicated a diagnosis of myxomas. Recurrence at the same site indicated that surgery was incomplete. In patient 2, the diagnosis of Carney complex cannot be ruled out due to recurrence of myxomas at different sites. In case 3, the diagnosis of Carney complex was established by the presence of lentigenes, pigmented cortical nodular hyperplasia, and a recurring left ventricular myxoma. Two of the patient's siblings also had altered skin pigmentation. In patient 4, Carney complex was diagnosed on the basis of a combination of myxomas at different sites and alterations in the PRKAR1A gene in the patient and the patient's mother. In patient 5, the diagnosis of Carney complex was based on the presence of lentiginosis, recurring myxomas, and genetic alterations in the patient and 2 of her children. Furthermore, in one of the children a myxoma was resected in the left atrium.
In our series, the Carney complex represented 4.7%-6.3% of cardiac myxomas. In addition to the observed recurrence, the myxomas were often multiple and located in any of the 4 cavities. However, as observed in one of patient 5's children, the classic location of a single tumor in the left atrium does not rule out the diagnosis. Cardiac myxomas develop earlier in this syndrome; patient 5's child developed the condition at age 12. Mean age in the group with recurrent myxoma was 32 years; in the group with isolated myxoma, mean age was 48 years.
Incidence of blood clots in the group with isolated atrial myxoma was 18%, which is similar to previously reported incidence rates.9 In the group with recurring myxomas, incidence was 40% and in patient 4 it is likely that the myocardial infarction was secondary to coronary embolism.
Carney complex follows an autosomal dominant inheritance pattern. Linkage analysis in families with this anomaly has shown genetic heterogeneity with at least 2 major loci as candidate genes. We initially identified a locus on chromosome 2 (2p15-16),10 but the gene that causes Carney complex has not been identified in this region. The PRKAR1A13 gene was recently identified at the second locus, located on chromosome 17 (17q22-24).11,12 This gene encodes the regulatory subunit of 1-alpha protein kinase A (PKA), the main mediator of cyclical adenosine phosphate signaling in mammals.14
Altered expressions of this gene have been identified in many sporadically occurring tumors and in tumor cell lines derived from patients without Carney complex, and it is thought to be a candidate gene for endocrine and non-endocrine tumorigenesis in Carney complex.15,16
In 2 of the 5 patients with recurrent myxomas, we found alterations in the PRKAR1A gene. The fact that we did not detect mutations in the PRKAR1A gene of individuals with isolated myxomas indicates that alterations of this gene are directly related to Carney complex and not with the emergence of myxomas. This is of clinical utility, as genetic alterations in patients with isolated myxomas at atypical sites, multiple myxomas, or myxomas at multiple sites could help to establish an early diagnosis of Carney complex. Once the diagnosis is established, genetic studies of family members can be performed and followed up by imaging studies in those who have the mutation. This in turn would allow for a program of clinical follow-up in patients and family members in whom mutations occur, given the risk of recurrence in those individuals. However, it is important to note that detecting mutations is not of use in cases where myxoma resection is incomplete, and that conventional methods will be required for detection in those cases. It should also be borne in mind that Carney complex is associated with genetically heterogeneous alterations, so that a lack of mutations in the PRKAR1A gene does not exclude the involvement of other genes; as mentioned earlier, the gene located on chromosome 2 may also be involved in producing Carney complex.
Limitations of the present study include the fact that it was observational and employed a retrospective design. The rarity of the disorders studied also meant that analysis was limited to a purely descriptive approach. The role of the genetic defect observed in patient 4 also remains to be determined and is the subject of an on-going study. The genetic study will be extended to the remaining 63 patients who were not included in the present analysis and who are attended in the outpatient department.
The results of the present study suggest the following conclusions: a) possible Carney complex should be investigated in all patients with intracardiac myxomas; b) Carney complex is more common in patients with recurrent or relapsing myxomas, and tumors are likely to occupy 2 or more cardiac cavities; and c) detecting mutations in patients with isolated myxomas at atypical sites, or with multiple myxomas, and screening for mutations in their immediate family, could be useful in establishing an early diagnosis of the disease and implementing appropriate clinical follow-up to detect recurrence in these individuals.
Correspondence: Dr. J. Vargas-Barrón.
Departamento de Ecocardiografía. Instituto Nacional de Cardiología Ignacio Chávez.
Juan Badiano, 1. Col. Sección XVI. 14080 Tlalpan. Mexico DF. Mexico.
E-mail: eco_vargas@terra.com.mx
Received September 7, 2007.
Accepted for publication January 22, 2008.