
Primary carnitine deficiency (PCD) is an autosomal recessive disorder with an incidence of 1 in 40,000 to 120,000 births. PCD is caused by mutations in the SLC22A5 gene, which encodes the plasmalemmal organic cation transporter type 2 (OCTN2), primarily expressed in the myocardium. These mutations lead to a reduction in plasma carnitine concentrations (<5μM; normal, 10-55μM) and an increase in urinary carnitine excretion, causing a decline in the transfer of long-chain fatty acids from the cytoplasm into the mitochondria.1
Most reported cases of PCD occur in infants and very young children. These patients often display multisystem involvement, including hepatomegaly, hypoketotic hypoglycemia, elevated ammonia levels in blood, organic acidosis, and hepatic encephalopathy. School-aged children (5-6 years) typically exhibit muscle weakness, elevated transaminases, and heart failure (hypertrophic or dilated cardiomyopathy).1,2
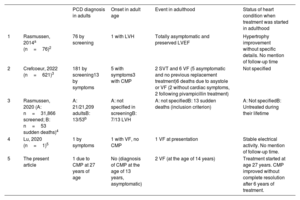
Onset is extremely rare in adolescence or adulthood. In these age groups, the diagnosis and specific treatment may be delayed for years, as cardiologists attending adults are not highly aware of this condition. A recent systematic review identified 161 patients with a symptom-based diagnosis of PCD. Of the 621 patients included, only 13 cases were in adults (table 1). The typical manifestations in adulthood are exclusively cardiac-related, commonly involving the triad of hypertrophic or dilated cardiomyopathy, noncompaction cardiomyopathy, and short QT interval. Without an accurate diagnosis and appropriate treatment, patients are at risk of ventricular arrhythmias and sudden cardiac death (23/621, 3.7% of symptomatic cases, 8 in adults) or severe heart failure.3,4,5
Summary of PCD cases in adults
| PCD diagnosis in adults | Onset in adult age | Event in adulthood | Status of heart condition when treatment was started in adulthood | ||
|---|---|---|---|---|---|
| 1 | Rasmussen, 2014a (n=76)2 | 76 by screening | 1 with LVH | Totally asymptomatic and preserved LVEF | Hypertrophy improvement without specific details. No mention of follow-up time |
| 2 | Crefcoeur, 2022 (n=621)3 | 181 by screening13 by symptoms | 5 with symptoms3 with CMP | 2 SVT and 6 VF (5 asymptomatic and no previous replacement treatment)6 deaths due to asystole or VF (2 without cardiac symptoms, 2 following pivampicillin treatment) | Not specified |
| 3 | Rasmussen, 2020 (A: n=31,866 screened; B: n=53 sudden deaths)4 | A: 21/21,209 adultsB: 13/53b | A: not specified in screeningB: 7/13 LVH | A: not specifiedB: 13 sudden deaths (inclusion criterion) | A: Not specifiedB: Untreated during their lifetime |
| 4 | Lu, 2020 (n=1)5 | 1 by symptoms | 1 with VF, no CMP | 1 VF at presentation | Stable electrical activity. No mention of follow-up time. |
| 5 | The present article | 1 due to CMP at 27 years of age | No (diagnosis of CMP at the age of 13 years, asymptomatic) | 2 VF (at the age of 14 years) | Treatment started at age 27 years. CMP improved without complete resolution after 6 years of treatment. |
CMP, cardiomyopathy; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; PCD, primary carnitine deficiency; SVT, sustained ventricular tachycardia; VF, ventricular fibrillation;
Regarding treatment, early supplementation with oral L-carnitine at a dose of 100 to 300mg/kg is effective in young children, with rapid and complete reversal of the phenotype. L-carnitine supplementation is safe at long-term, with reported follow-ups of more than 30 years. Even after cardiologic normalization, interrupting this treatment may lead to clinical deterioration, which can be reverted if supplements are resumed.2,3 There is considerable evidence supporting the benefits of early treatment, but a striking lack of information on the outcome in adults starting treatment many years after receiving the cardiomyopathy diagnosis.
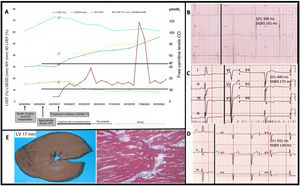
We present the case of a 27-year-old man diagnosed with PCD after years of symptomatic treatment for dilated cardiomyopathy with hypertrophy, noncompaction, and short QT interval. After the sudden death of his sister, the patient started regular follow-up visits, including 2 genetic studies that yielded no relevant findings. A short time later, the patient underwent defibrillator implantation for primary prevention, which delivered 2 appropriate therapies for ventricular fibrillation at 6 months. Fourteen years later, the diagnosis of PCD was established based on clinical manifestations (low carnitine levels) and genetic criteria (compound heterozygote for the pathogenic SLC22A5 variants p.Glu452Lys and p.Leu269HisfsTer27). Weight-adjusted L-carnitine replacement treatment was started (commercial brand followed by compounded formulations, initially in dry powder form and later as a syrup), which achieved relatively stable carnitine values within the normal range. The QT interval improved rapidly, and no further arrhythmic events were detected. Systolic function and ventricular dimensions have improved, but not completely normalized, in contrast to the response reported for treatment started promptly and in childhood (figure 1). The noncompaction has remained unchanged. The patient's parents and 1 healthy sibling were identified as simple heterozygotes.

A: PCD patient starting carnitine replacement treatment at the age of 27 years, after a 14-year delay since onset of heart disease. B–D: electrocardiograms at the time of diagnosis, shortly after onset, and following 5 years of treatment. E: images from autopsy study of the patient's sister: macroscopic view shows LV hypertrophy, and histological image reveals areas of disarray.
Diagnosis and treatment of PCD in adults poses a series of challenges that will be discussed. A presymptomatic diagnosis is often achieved through systematic neonatal screening for endocrine and metabolic diseases. However, when many of the current adult patients were born, PCD was not included in these programs. Moreover, PCD screening is not consistently performed across regions and has recognized pros and cons.
PCD is largely unfamiliar to cardiologists. Hence, it may be treated simply as an underlying cardiomyopathy, without the specific supplementation that can reverse or mitigate the phenotype, and without appropriate family-centered management.
Several commercial genetic panels do not include the SLC22A5 gene, and others incorporated it only between 2015 and 2020. Therefore, SLC22A5 was not investigated in older studies. Following diagnosis, adults require high, weight-adjusted doses of L-carnitine, which often leads to a common side effect: strong body odor resembling decayed fish.
Regular monitoring is required to ensure that the free carnitine concentration stays within the normal range, even when treatment adherence is adequate and there are no signs of clinical decompensation.
Reported information is scarce on the safety and effectiveness of carnitine replacement initiated many years after onset of the cardiomyopathic phenotype. In response to the aforementioned challenges, the authors suggest the following:
- •
It is important to remember that some undiagnosed patients without neonatal screening may reach adulthood and present with heart disease.
- •
Cardiologists attending adults should become more aware of PCD to better suspect and confirm the presence of this condition.
- •
Advances in cardiogenetic research, with an increasing number of genes identified in exome and genome studies, will enable systematic analysis of minority genes such as SLC22A5. When gene panels are used, health care providers should be aware of the clinical triad of hypertrophic or dilated cardiomyopathy, noncompaction, and short QT interval to purposely include the SLC22A5 gene in the analysis.
- •
The high life-long doses of L-carnitine required in adolescents and adults suggest a treatment strategy that avoids commercial brands (need for many vials and high cost). A better option would be more concentrated formulas compounded in hospital pharmacies in dry powder or syrup form. In our experience, syrup formulations have proved effective for achieving stable values within the normal range.
- •
Unpleasant body odor caused by intestinal bacteria overgrowth can be treated with a 10mg/kg/day dose of metronidazole for 10 days each month.6
- •
To facilitate follow-up, remote appointments can be scheduled between in-person visits. In this approach, patients send 3 blood drops on Whatman paper by mail directly to the laboratory for free carnitine analysis.
Based on our experience and a review of the literature, we have outlined some useful suggestions to improve the diagnosis and treatment of PCD patients diagnosed in adulthood. It is important to note that the prospect of complete cure is not as favorable in this population as in young children, especially when the start of carnitine treatment is delayed for years after development of the cardiac phenotype.
FUNDINGThe present research has not received specific financial support from either the private or public sector.
ETHICAL CONSIDERATIONSThis study was approved by the Ethics Committee of Hospital Universitario y Politécnico La Fe de Valencia. Informed consent was obtained from patients for publication of their case. This study adheres to the SAGER guidelines.
STATEMENT ON THE USE OF ARTIFICIAL INTELLIGENCEArtificial intelligence was not used in the development of the present study.
AUTHORS’ CONTRIBUTIONSAll authors contributed, to a greater or lesser extent, to all the following tasks: study conception and design; data acquisition, analysis, and interpretation; drafting the article and critically revising the intellectual content; and approving the version to be published. All authors agree to take responsibility for all aspects of the article and to investigate and resolve any issues related to the accuracy of its parts.
CONFLICTS OF INTERESTNone.
We greatly appreciate the participation of the patient and his family, the Sevicio de Análisis Clínicos-Metabolopatías of our center, and the Instituto de Medicina Legal de Valencia. The samples included were handled by Biobanco La Fe (B.0000723) and sequenced in the Unidad de Genómica del Instituto de Investigación Sanitaria La Fe de Valencia.