
Cardiorenal syndrome has been defined as the simultaneous dysfunction of both the heart and the kidney. Worsening renal function that occurs in patients with acute heart failure has been classified as cardiorenal syndrome type 1. In this setting, worsening renal function is a common finding and is due to complex, multifactorial, and not fully understood processes involving hemodynamic (renal arterial hypoperfusion and renal venous congestion) and nonhemodynamic factors. Traditionally, worsening renal function has been associated with worse outcomes, but recent findings have revealed mixed and heterogeneous results, perhaps suggesting that the same phenotype represents a diversity of pathophysiological and clinical situations. Interpreting the magnitude and chronology of renal changes together with baseline renal function, fluid overload status, and clinical response to therapy might help clinicians to unravel the clinical meaning of renal function changes that occur during an episode of heart failure decompensation. In this article, we critically review the contemporary evidence on the pathophysiology and clinical aspects of worsening renal function in acute heart failure.
Keywords
Heart failure (HF) is the first cause of hospitalization in persons aged 65 years or older and represents a substantial percentage of all hospital admissions and health care costs.1 Heart failure is a complex syndrome that affects almost all organs and systems of the body. Renal dysfunction is one of the most important comorbidities in patients with chronic HF and is accentuated, or becomes more evident, during episodes of acute heart failure (AHF).2–4 The association between the heart and the kidney in patients with AHF is complex, and a complete understanding of this 2-directional interaction has not been elucidated.5,6 In the AHF setting, worsening renal function (WRF) is a prevalent condition ranging from 10% to 40% of patients. Despite its high prevalence, WRF still represents a diagnostic, prognostic, and therapeutic challenge.2–4 In this article, we critically review the pathophysiology and clinical implications of WRF in AHF, especially by relating the new information to old paradigms.
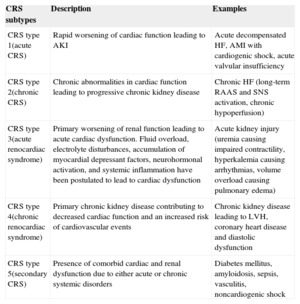
DEFINITIONCardiorenal syndrome (CRS) has been defined as the simultaneous dysfunction of both the heart and the kidney, regardless of which of the 2 organs suffered the initial damage and their previous functional status.6 This syndrome has been classified in an academic manner by the Acute Dialysis Quality Initiative working group, which proposes a classification scheme of 5 subtypes.5,6 This 5-item classification is based on: a) whether the primary organ of dysfunction is the heart, the kidney, or a third independent process affecting both organs, and b) the acute or chronic nature of the disease. Table 1 summarizes the current 5 CRS subtypes.
Classification and Definition of Cardiorenal Syndrome
| CRS subtypes | Description | Examples |
|---|---|---|
| CRS type 1(acute CRS) | Rapid worsening of cardiac function leading to AKI | Acute decompensated HF, AMI with cardiogenic shock, acute valvular insufficiency |
| CRS type 2(chronic CRS) | Chronic abnormalities in cardiac function leading to progressive chronic kidney disease | Chronic HF (long-term RAAS and SNS activation, chronic hypoperfusion) |
| CRS type 3(acute renocardiac syndrome) | Primary worsening of renal function leading to acute cardiac dysfunction. Fluid overload, electrolyte disturbances, accumulation of myocardial depressant factors, neurohormonal activation, and systemic inflammation have been postulated to lead to cardiac dysfunction | Acute kidney injury (uremia causing impaired contractility, hyperkalemia causing arrhythmias, volume overload causing pulmonary edema) |
| CRS type 4(chronic renocardiac syndrome) | Primary chronic kidney disease contributing to decreased cardiac function and an increased risk of cardiovascular events | Chronic kidney disease leading to LVH, coronary heart disease and diastolic dysfunction |
| CRS type 5(secondary CRS) | Presence of comorbid cardiac and renal dysfunction due to either acute or chronic systemic disorders | Diabetes mellitus, amyloidosis, sepsis, vasculitis, noncardiogenic shock |
AKI, acute kidney injury; AMI, acute myocardial infarction; CRS, cardiorenal syndrome; HF, heart failure; LVH, left ventricular hypertrophy; RAAS, renin-angiotensin-aldosterone system; SNS, sympathetic nervous system.
Although this classification is a step forward in the search to understand the complexity of cardiorenal interaction, it is not easily applied in clinical practice. Thus, further attempts to better dissect and categorize CRS are warranted. In the following paragraphs, we will focus on WRF occurring in the setting of AHF decompensation of chronic HF or as de novo HF (CRS type 1).
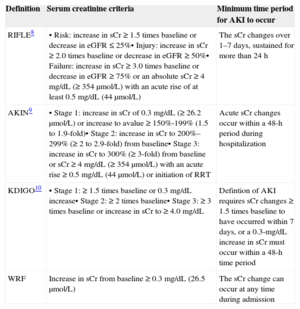
Traditionally, acute kidney injury (AKI) in AHF patients is defined by WRF during hospitalization, which has been broadly defined as serum creatinine changes ranging from ≥ 0.3 to 0.5mg/dL.4 Nevertheless, this definition lacked universal consensus.7 Indeed, there appears to be a notorious disagreement between HF and nephrology guidelines on the best criteria for WRF.7–10
Moreover, current AKI definitions have been validated mostly in non-HF scenarios. Table 2 summarizes different current AKI definitions including RIFLE8 (Risk, Injury, Failure, Loss of Kidney Function, and End-stage Kidney Disease), AKIN9, KDIGO10, and WRF criteria.
| Definition | Serum creatinine criteria | Minimum time period for AKI to occur |
|---|---|---|
| RIFLE8 | • Risk: increase in sCr ≥ 1.5 times baseline or decrease in eGFR ≤ 25%• Injury: increase in sCr ≥ 2.0 times baseline or decrease in eGFR ≥ 50%• Failure: increase in sCr ≥ 3.0 times baseline or decrease in eGFR ≥ 75% or an absolute sCr ≥ 4 mg/dL (≥ 354μmol/L) with an acute rise of at least 0.5 mg/dL (44μmol/L) | The sCr changes over 1–7 days, sustained for more than 24 h |
| AKIN9 | • Stage 1: increase in sCr of 0.3 mg/dL (≥ 26.2 μmol/L) or increase to avalue ≥ 150%-199% (1.5 to 1.9-fold)• Stage 2: increase in sCr to 200%–299% (≥ 2 to 2.9-fold) from baseline• Stage 3: increase in sCr to 300% (≥ 3-fold) from baseline or sCr ≥ 4 mg/dL (≥ 354μmol/L) with an acute rise ≥ 0.5 mg/dL (44μmol/L) or initiation of RRT | Acute sCr changes occur within a 48-h period during hospitalization |
| KDIGO10 | • Stage 1: ≥ 1.5 times baseline or 0.3 mg/dL increase• Stage 2: ≥ 2 times baseline• Stage 3: ≥ 3 times baseline or increase in sCr to ≥ 4.0 mg/dL | Defintion of AKI requires sCr changes ≥ 1.5 times baseline to have occurred within 7 days, or a 0.3-mg/dL increase in sCr must occur within a 48-h time period |
| WRF | Increase in sCr from baseline ≥ 0.3 mg/dL (26.5μmol/L) | The sCr change can occur at any time during admission |
AKI, acute kidney injury; AKIN, Acute Kidney Injury Network; eGFR, estimated glomerular filtration rate; KDIGO, Kidney Disease: Improving Global Outcomes; RIFLE, Risk, Injury, Failure, Loss of Kidney Function, and End-stage Kidney Disease; RRT, renal replacement therapy; sCr, serum creatinine; WRF, worsening wenal function.
Perhaps fueling this discrepancy in the criteria for AKI in AHF is the underlying complex and multifactorial pathophysiology pathways involved and the different patient populations used by the different studies. All of these factors together explain the most important differences in the epidemiology and clinical implications of renal function changes observed among different studies (Table 3).11–28
Definition, Incidence and Prognostic Implications of Worsening Renal Function in Acute Heart Failure
| Authors | WRF definition | Incidence of WRF | Conclusions |
|---|---|---|---|
| Mullens et al11 | Increase ≥ 0.3 mg/dL in sCr | 40% of patients admitted for AHF | Venous congestion is the most important hemodynamic factor driving WRF in decompensated patients with advanced HF |
| Metra et al12 | Occurrence of both an increase ≥ 25% or ≥ 0.3 mg/dL in sCr | 34% of patients admitted for AHF | WRF is a frequent finding in patients hospitalized for AHF and is associated with poor prognosis. Severity of HF and daily furosemide dose are the most important predictors of the occurrence of WRF |
| Damman et al13 | Increase > 26.5μmol/L and > 25% in sCr | In-hospital WRF occurred in 11% of patients, while 16% and 9% experienced WRF from 0 to 6, and 6 to 12 months after discharge, respectively | Both in- and out-hospital WRF are independently related to poor prognosis in patients with HF, suggesting that renal function in HF patients should be monitored long after discharge |
| Forman et al14 | Increase > 0.3 mg/dL in sCr (26.5μmol/L) | 27% of patients admitted for AHF | WRF occurs frequently among hospitalized HF patients and is associated with significantly worse outcomes. Clinical characteristics available at hospital admission can be used to identify patients at increased risk for developing WRF |
| Voors et al15 | Increase ≥ 0.3 mg/dL in sCr by day 5 from admission. | 30% of patients admitted for AHF | Worsening renal function in hospitalized AHF patients is related to a poor clinical outcome and is predicted by a greater early drop in SBP |
| Akhter et al16 | Increase ≥ 0.5 mg/dL in sCr | 24.8% of patients admitted for AHF | An increase in sCr in the hospital results in a significantly longer length of stay and has an independent effect on long-term mortality |
| Chittineni et al17 | Increase of 0.5 mg/dL in sCr | 21% of patients admitted for AHF | ARF is a common complication among patients hospitalized for congestive HF, and is associated with increased risk for adverse outcomes. Certain clinical characteristics present at the time of admission help identify patients at increased risk |
| Gottlieb et al18 | Various definitions of worsening renal function | 72% of patients developedincreased sCr during hospitalization,with 20% developing an increaseof ≥ 0.5 mg/dL | This analysis demonstrates that any detectable decrease in renal function is associated with increased mortality and prolonged hospital stay. This suggests that therapeutic interventions which improve renal function might be beneficial |
| Aronson and Burger19 | Increase ≥ 0.5 mg/dL in sCr above baseline at any time pointPersistent when sCr remained ≥ 0.5 mg/dL above baseline throughout day 30, and transient when sCr levels subsequently decreased to < 0.5 mg/dL above baseline | WRF occurred in 115 patients (n=467), and was transient in 39 patients (33.9%) | Transient WRF is frequent among patients with AHF. Whereas persistent WRF portends increased mortality, transient WRF appears to be associated with a better outcome as compared with persistent renal failure |
| Cowie et al20 | Increase in sCr >26μmol/L (approximately 0.3 mg/dL) from admission | 29% of patients admitted for AHF with a history of LVEF ≤ 40% | WRF is common in patients admitted to European hospitals with decompensated HF. Such patients have longer admissions, but a similar mortality and rehospitalization rate to those without WRF (if patients experiencing a major in-hospital complication are excluded) |
| Blair et al21 | Increase in sCr ≥ 0.3 mg/dL during the in-hospital (randomization to discharge or day 7) and postdischarge (discharge or day 7 to 4 weeks postdischarge) periods | 13.8% in-hospital patients and 11.9% postdischarge patients with reduced LVEF (≤ 40%) | The prevalence of renal dysfunction is high in patients hospitalized for HF. Worsening renal function may occur not only during hospitalization, but also in the early postdischarge period. Since worsening renal function during hospitalization is associated with a significant decrease in signs and symptoms of congestion, body weight and natriuretic peptides, which are good prognostic indicators, worsening renal function during hospitalization as an endpoint in clinical trials should be reevaluated |
| Krumholz et al22 | Increase > 0.3 mg/dL in sCr during hospitalization | 28% of patients admitted for AHF | WRF, an event that frequently occurs in elderly patients hospitalized with HF, confers a substantial burden to patients and the healthcare system and can be predicted by 6 admission characteristics |
| Kociol et al23 | Increase ≥ 0.3 mg/dL in sCr | 17.8% of patients ≥ 65 years of age hospitalized with HF and discharged alive | WRF in patients hospitalized with HF was independently associated with long-term mortality |
| Damman et al24 | Increase ≥ 0.2 mg/dL in sCr or a corresponding decrease in eGFR ≥ 5mL × min × 1.73 m2 | WRF developed in 25% patients | WRF predicts substantially higher rates of mortality and hospitalization in patients with HF |
| Klein et al25 | Decrease >25% in eGFR or increase > 25% in BUN | By discharge, 12% of patients had a > 25% decrease in eGFR, and 39% had a > 25% increase in BUN | A substantial number of patients admitted with heart failure have worsening renal function during hospitalization. Higher admission BUN and increasing BUN during hospitalization, independently of admission values, are associated with a worse survival rate. Use of milrinone in these high-risk patients does not improve outcomes despite minor improvements in renal function |
| Testani et al26 | IRF was defined as a ≥ 20% improvement and WRF as a ≥ 20% deterioration in eGFR | 31.4% of patients experienced IRF | IRF is associated with significantly worsened survival and may represent the resolution of venous congestion-induced preadmission WRF. Unlike WRF, the renal dysfunction in IRF patients occurs independently of the confounding effects of acute decongestion and may provide incremental information for the study of cardiorenal interactions |
| Smith et al27 | Different WRF definitions (absolute creatinine elevations ≥ 0.1 to ≥ 0.5 mg/dL and 25% relative elevation from baseline) | sCr elevation ≥ 0.1 mg/dL occurred in 75% of patients, and elevation ≥ 0.5 mg/dL occurred in 24% of patients | Larger creatinine elevations predict the highest risk of death, yet even minor changes in renal function are associated with adverse outcomes. The choice of a “best definition” for WRF has implications for the number of patients identified with this risk factor and the magnitude of risk for mortality |
| Belziti et al28 | Increase > 0.3 mg/dL in sCr and additionally, by at least 25% with respect to the baseline value during hospitalization | 23% of patients experienced WRF | WRF is a common complication in AHF patients and is associated with a longer hospital stay and an increased risk of mortality or HF readmissions |
AHF, acute heart failure; ARF, acute renal failure; BUN, blood urea nitrogen; eGFR, estimated glomerular filtration rate; HF, heart failure; IRF, improvement renal function; LVEF, left ventricular ejection fraction; SBP, systolic blood pressure; sCr, serum creatinine; WRF, worsening renal function.
Chronic renal failure is extremely common in patients with HF, with a prevalence ranging from 20% to 57% in chronic stable HF patients and 30% to 67% in large registries of AHF.29 Among those admitted for AHF, the occurrence of WRF ranged between 10% and 40%.2–4,11–28 As previously mentioned, this wide variability can be attributed, at least in part, to different cut-off values used to define WRF, differences in the time at risk, different characteristics of the study population, and the varying degrees of diagnostic accuracy of the available methods.
Several risk factors have been associated with the development of WRF, including age,13,15,17 male sex,13 prior known renal insufficiency,12–15,17–19,21,22,28 diabetes mellitus,13,14,17,22 a prior history of HF,30 a prior history of WRF,31 high and low systolic blood pressure, a significant drop in systolic blood pressure,13–15,22 atrial fibrillation,20 low serum sodium,17 diastolic dysfunction,17 pulmonary edema,13,20 furosemide dose,12 or sequential nephron blockade with the combination of loop diuretics and thiazides.32 Of note, left ventricular ejection fraction is not a well-established risk factor for developing WRF.33
PATHOPHYSIOLOGYThe pathophysiological mechanisms responsible for CRS type 1 are complex, multifactorial and not entirely understood. An imbalance between abnormal hemodynamics, neurohormonal activation, inflammatory responses, intrinsic tubular damage, and heterogeneous response to therapeutic interventions have been postulated.6,34
Hemodynamic MechanismsLow Cardiac Output-renal HypoperfusionSeveral experimental and clinical data indicate that hemodynamics play a major role, if not the most important one, in the pathophysiology of CRS type 1. Traditionally, WRF has been attributed to hypoperfusion of the kidney due to low cardiac output.14 Reduced cardiac output and central fluid redistribution portend decreased renal perfusion. As compensatory mechanisms, stimulation of the sympathetic nervous system, renin-angiotensin-aldosterone system, and vasopressin secretion lead to enhanced water and sodium reabsorption, in an effort to preserve renal perfusion and glomerular filtration rate (GFR); however, in the long-term, this type of response induces deleterious effects in the heart and kidney by promoting fibrosis, apoptosis, and ventricular remodeling.35 Furthermore, persistent hypoperfusion may even lead to renal parenchyma/cortical ischemia, which by itself, further compromises renal function.35
Contemporary thinking recognizes that low cardiac output can explain only a minor part of the pathogenesis of CRS type 1, and it appears not to be the primary determinant of WRF in daily clinical practice. In large registries, the proportion of patients with a “cold” profile or hypotension at admission is relatively small.36,37 Likewise, important drops in systolic blood pressure related to WRF are not frequently observed in clinical daily practice.15 In an attempt to endorse previous observations, the results from the ESCAPE trial38 showed no correlation between WRF and cardiac index or systemic vascular resistances. Similarly, Mullens et al11 reported that patients who developed WRF did not have a lower cardiac index on admission than those without WRF. Collectively, most of the current evidence does not support low cardiac output as the main determinant of WRF in patients with AHF syndromes.
Fluid Overload-renal Venous CongestionDecades ago, experimental researchers demonstrated that temporary isolated elevation of central venous pressure (CVP) decreased renal blood flow and GFR.39,40 Winton observed that diuresis by an isolated canine kidney was markedly reduced at a renal venous pressure of 20mmHg and was abolished at pressures > 25mmHg.40 In an early experiment in normal individuals, reaching an intra-abdominal pressure of 20mmHg with abdominal compression markedly reduced GFR.41 Recent studies have translated this historical experimental data into current clinical practice, reporting an association between high venous pressures and WRF, which seems to be superior to the effect of arterial blood pressure, cardiac index, or pulmonary capillary wedge pressure as predictors for CRS type 1.11,42,43 The mechanisms postulated to explain these findings include: a) increased pressure along renal veins reduces the net pressure gradient across the glomerulus, decreasing GFR; b) the resultant increased renal interstitial pressure may lead to tubular compression, parenchymal hypoxia,3 and additional reduction in GFR, and; c) extrinsic compression (eg, abdominal hypertension) of renal veins and parenchyma has also been shown to impair renal function.3,5,6,11,42,43
However, high venous pressure-related WRF has not been a consistent finding38,44,45 and other experimental and clinical studies suggest that elevations in CVP become highly relevant, especially in conditions with marked abnormal hemodynamics. In an animal model of renal venous hypertension, GFR only declined when cardiac output was concomitantly impaired.46 Similarly, recent clinical studies reported that CVP was an independent predictor of WRF, but especially when there was low cardiac output.11,44 These apparently conflicting results may reflect the multifactorial nature of this interaction. Of note, it must be stressed that CVP is not a reliable surrogate of fluid overload because, in the venous pressure system, it has little correlation with volume.47 The high compliance of the venous system facilitates a relative pressure-volume disconnection, so large changes in volume are associated with only small pressure changes. In addition, venous pressure is driven by the combination of volume and venous tone. Venous tone is primary mediated by neurohormonal activation, so CVP not only depends on volume, but also on the triggered systemic neurohormonal response.47 This might explain why some authors have failed to show significant correlations between CVP and measures of volume status,48 and why right atrial pressure was not a reliable surrogate of the magnitude of decongestion in AHF and thus a poor predictor of WRF risk.49 Although there are no reliable surrogates of systemic congestion, bioelectrical impedance or biomarkers such as carbohydrate antigen 125 have been tested with encouraging results.50,51 Further studies are definitely needed in this field. To better characterize the role of congestion in the CRS pathophysiology, a comprehensive evaluation of hydration status must be followed when patients are admitted for AHF.
Neurohormonal Activation and Sympathetic ActivitySeveral neurohormonal and inflammatory pathways are implicated in the pathophysiology of renal dysfunction in AHF.35 Renin-angiotensin-aldosterone system activation portends to maintain GFR in acute hypoperfusion situations; nevertheless, persistent stimulation plays a key role in kidney damage through cell hypertrophy, fibrosis stimulation, oxidative stress, and activation of inflammatory mechanisms.52 Angiotensin II is a potent systemic vasoconstrictor that promotes arteriolar constriction, decreasing renal blood flow, and stimulates the sympathetic nervous system. The sympathetic nervous system increases systemic vascular tone and has direct untoward effects in the heart and kidney by promoting apoptosis and fibrosis.52 Stimulation of adrenergic receptors on proximal tubular cells enhances sodium reabsorption, whereas adrenergic receptors in the juxtaglomerular apparatus further stimulate the renin-angiotensin-aldosterone system. Aldosterone secretion leads to salt and water retention, thus contributing to edema and congestion.6,52 In addition, an intrarenal renin-angiotensin-aldosterone system neurohormonal component has also been described that modulates renal function intrinsically.3
Furthermore, sympathetic activity is heightened as a recognized precipitant factor in HF decompensation, which is reflected in the finding that redistribution of intravascular volume rather than a change in total salt or water is an important driving force, with neither an increase in total body fluid nor previous weight gain.6,53 For instance, Fallick et al53 claim that acute changes in vascular splanchnic venous capacitance regulated by sympathetic tone can lead to an abrupt translocation of volume to the effective circulatory bench, leading to acute central venous hypertension.
Inflammatory ResponseSeveral studies support the concept of HF as an immune dysregulation scenario.54 Elevations of cytokines and other markers of inflammation have been documented in AHF patients.55 Inflammatory cytokines, such as tissue necrosis factor-α, have been proposed to play a role in sodium retention, myocardial dysfunction, AKI, vascular dysfunction, and extracellular fluid overload.6 In addition, inflammation seems to be largely associated with inadequate renal perfusion pressures, peritubular edema, pathological reduction of glomerular filtration, and tubular damage (on top of the effect of ischemia).6 In an experimental human model of venous congestion, Colombo et al56 recently showed that, in normal individuals, peripheral venous congestion per se causes release of inflammatory mediators, neurohormones, and activation of endothelial cells.
Intrinsic Tubular DamageDifferent mechanisms have been proposed for the development of intrinsic tubular damage in AHF syndromes. The most important are probably decreased local perfusion and high venous pressures, leading to ischemia and high intrarenal interstitial pressures.57 In recent years, new sensitive and specific markers of tubular damage, such as neutrophil gelatinase-associated lipocalin (NGAL) have been explored in HF, showing that this tubular injury marker appears in urine and plasma long before serum creatinine increase.58 In a cohort of 2011 chronic HF patients, Damman et al59 recently showed that tubular damage markers are strongly associated with WRF risk. The presence of tubular injury markers in the chronic setting could reflect chronic renal hypoxia and enhanced vulnerability to the hemodynamic changes and neurohormonal responses that arise in HF decompensations. How these episodes of AKI can damage nephron units, leading to future renal dysfunction and/or adverse outcomes, is still poorly understood.
Therapeutic InterventionsLoop diuretics are used almost universally to relieve congestion and improve symptoms in HF, and are still the cornerstone of treatment during HF decompensations. There are, however, some concerns about their safety profile because of the association with deleterious neurohormonal activation, renal dysfunction, and even poor clinical outcomes.6,60 In AHF, individual clinical response to diuretics and their effect on renal function are markedly heterogeneous.61 Worsening renal function induced by intensive diuretic treatment may be the result of several pathophysiological and clinical situations. In fact, it has been suggested that this double edged-sword of the effect of loop diuretics on renal function is largely determined by a delicate balance between renal perfusion and venous congestion.62,63 On the harmful side, they can lead to intravascular volume depletion, reduced renal perfusion and deterioration in renal function. On the beneficial side, loop diuretics can decrease venous congestion, and therefore, improve GFR.62,63
In addition, recent studies have suggested that, at least in some patients, WRF might be a surrogate for hemoconcentration after aggressive decongestion, and associated, at least temporary, with better outcomes.64–67In the DOSE (Diuretic Optimization Strategies Evaluation) trial, a transient WRF with the use of high-dose diuretics was associated with early clinical improvement and not worse prognosis at 60 days.65 In 599 consecutive patients with AHF, Metra et al66 found that the prognostic value of WRF was mainly determined by the presence of congestion; in the absence of congestion, increases in serum creatinine levels had no prognostic value; in contrast, WRF was strongly associated with a higher risk of adverse outcomes in patients with persistent congestion. Similarly, in an analysis of the ESCAPE trial, Testani et al67 showed that hemoconcentration was associated with WRF and with better outcomes. These data suggest that high-dose diuretics are beneficial in a volume overload status, but can be hazardous in patients with mild fluid overload or fluid redistribution. In summary, this bimodal effect of diuretics on renal function emphasizes the heterogeneity of AHF syndromes and highlights the importance of accurate assessment of fluid overload for tailoring the diuretic dose.
CLINICAL IMPLICATIONSDiagnosisCreatinine and UreaTraditional markers of renal function, such as serum creatinine or blood urea nitrogen, have been classically used as surrogates for renal function, but there are several concerns regarding their performance, especially in HF decompensations.64–69 Serum creatinine is almost universally used but is influenced by important extrarenal factors such as muscle mass, sex, age, and race. Serum creatinine underestimates renal function in older persons and women and in low-weight individuals, a profile commonly found in patients with AHF. In contrast, creatinine changes overestimate renal damage when renal dysfunction is already present.70 In addition, creatinine is known to be a slow-releases marker in AKI (it is increased up to 24h after renal injury), which constitutes another important limitation.69 As mentioned before, an increase in creatinine may occur as a consequence of hemoconcentration, even in the absence of any renal damage, as it often occurs in patients with AHF treated with intensive diuretic therapy.64–67
Similarly, urea levels are also substantially influenced by neurohormonal activation, protein intake, and catabolic processes. Activation of the renin-angiotensin-aldosterone system increases urea reabsorption in the proximal tubule, a process that is linked to sodium and water reabsorption, whereas vasopressin levels enhance reuptake in the collecting duct, through activation of urea transporters.29 Thus, urea levels reflect persistent and inappropriate renin-angiotensin-aldosterone system and vasopressin activation in HF, but are not necessarily related to a decrease in GFR.71 These shortcomings in the performance of classic renal function markers have stimulated the search for more specific and accurate surrogates of glomerular function, such as cystatin C, and new GFR formulas, such as the Modification of Diet in Renal Disease Study or the Chronic Kidney Disease Epidemiology Collaboration, which provide a more accurate assessment of renal function in HF.70
Novel BiomarkersGlomerular Damage. Cystatin C is a 122-aminoacid, 13-kDa, member of the family of cysteine proteinase inhibitors, produced by all nucleated cells at a constant rate, and has emerged as a marker of glomerular damage. Its superiority over other markers of renal function lies in the fact that it is freely filtered by the glomerulus and not secreted, although is slightly reabsorbed by tubular cells, where it is catabolized. Unlike creatinine and blood urea nitrogen, it is independent of muscle mass, protein intake or catabolism, which is why it has been postulated as a more specific and accurate marker of GFR.52 In AHF, its values at admission have been shown to be independently associated with mortality and readmissions.72 Interestingly, recent evidence supports the long-term prognostic utility of this biomarker beyond GFR in HF patients with moderate renal dysfunction (GFR, 30-60mL/min/1.73 m2).73 Additional data are warranted regarding the clinical value of cystatin C kinetics in patients with AHF.
Tubular Damage Markers. The clinical need for a more accurate and an early diagnosis of AKI has driven research to explore novel biomarkers related to tubulointerstitial injury, such as NGAL, tubular kidney injury molecule-1, and N-acetyl-β-D-glucosaminidase, capable of affording an early diagnosis of tubular damage in different clinical scenarios.52 Neutrophil gelatinase-associated lipocalin is a 178-aminoacid protein that belongs to the lipocalin family of proteins. In normal circumstances, only small amounts of NGAL can be found in plasma and urine. However, in response to AKI, NGAL is rapidly released (within 2h), increasing its levels dramatically.52 The usefulness of NGAL for diagnosing AKI and as a prognostic marker has been highlighted in a meta-analysis.74 Neutrophil gelatinase-associated lipocalin has been shown to be a more sensitive and specific marker than creatinine for the diagnosis of AKI in different scenarios, including AHF.58,75 In addition, serum, and also urinary NGAL, have been strongly related to death or readmission in AHF and chronic HF, an added value that is beyond natriuretic peptides and other renal indices.76–78
The tubular kidney injury molecule-1 is a type 1 transmembrane glycoprotein that mediates the conversion of cells into phagocytes and plays a role in the immune response to injury. It is a novel marker of proximal tubular damage,59 is measured in urine, and is only present in pathological conditions. In a cohort of 2011 chronic HF patients, Damman et al59 recently showed that tubular kidney injury molecule-1 was the strongest tubular marker in WRF prediction, superior to NGAL or N-acetyl-β-D-glucosaminidase, and moreover, patients with increased urinary tubular kidney injury molecule-1 levels have a significantly faster decline in GFR over time.
N-acetyl-β-D-glucosaminidase is a lysosomal brush border enzyme released by renal tubular proximal cells into urine after tubular injury.52 Along with tubular kidney injury molecule-1, it can only be measured in urine and is associated with adverse outcomes, independently of GFR.59 However, although both tubular kidney injury molecule-1 and N-acetyl-β-D-glucosaminidase have been evaluated in various acute clinical conditions, there is still no solid evidence about its performance in the setting of HF decompensations. Other future biomarkers under investigation, and potentially related to CSR type 1, include interleukin-18, liver-type fatty acid binding protein, osteopontin, stromal cell-derived factor 1, galectin 3, endoglin, and exosomes.
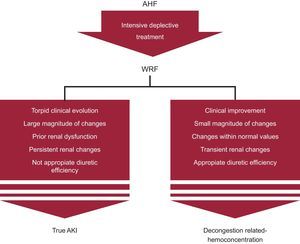
Risk AssessmentClassically, developing WRF in the setting of chronic HF and AHF has been shown to increase the prognostic burden of this disease.2–4 Overall, WRF in the setting of AHF has been related to higher hospitalization costs, longer hospital stay, and worse outcomes.2,4,29 Most relevant studies and their prognostic findings are summarized in Table 3. A recent meta-analysis of 28 studies (49 890 patients with AHF) reported that 23% of patients had WRF, as defined in the individual studies (which varied study by study).4 After a mean (standard deviation) follow-up of 418 (594) days, WRF was related to an increased risk of mortality (odds ratio, 1.75, 95% confidence interval, 1.47–2.08; P<.001), but this effect was more pronounced in patients with chronic HF (odds ratio, 1.96, 95% confidence interval, 1.48–2.61; P < .001).4 In this study, the authors pointed out that there was wide heterogeneity among results, reflecting the complexity of AHF syndromes and the different criteria used in the definition of WRF. Along this line, the most important challenge remains to distinguish between true AKI vs decongestion-related hemoconcentration (Figure). In an attempt to help clinicians to interpret renal function changes, we suggest taking into account the following characteristics:

Clinical course. When evaluating renal function changes, it is mandatory to adopt a comprehensive approach, considering symptoms, vital signs, and volume of diuresis, among others.
Fluid overload status. Recent observations have shown that WRF had no relationship with outcomes when occurring in patients with concomitant hemoconcentration.64–67 Thus, in hyperhydrated patients undergoing intensive diuretic therapy, WRF may be a proxy for decongestion and, as such, an indicator for appropriate response to treatment, and potentially, for better outcomes.64–67 Conversely, WRF occurring in the setting of persistent signs of congestion or in patients with presumable renal hypoperfusion might portend a worse prognosis.15,44,63–67
Baseline renal function and magnitude of renal changes. Recent observations have found that the clinical impact of changes in renal function within the first 48hours to 72hours after admission for AHF is largely determined by the presence of renal failure on admission and the magnitude of changes.79 Indeed, in patients with renal failure on admission, the increase in creatinine was independently associated with a higher risk of 1-year mortality and the excess of risk became significant with even small increases. In contrast, in patients with normal or mildly impaired renal function on admission, small creatinine changes seen in daily practice were not significantly associated with mortality and only important creatinine changes (greater than 1mg) were related to worse outcomes.79 In agreement with these results, the RELAX-AHF trial, which included patients with renal dysfunction, a creatinine increase ≥ 0.3mg/dL and cystatin-C increase ≥ 0.3mg/L at for 2 days were associated with a higher risk of 180-day mortality (hazard ratio = 1.76; 95% confidence interval, 1.11–2.82 and hazard ratio = 2.10; 95% confidence interval, 1.38-3.20, respectively).80 We postulate that small changes in patients without established renal failure represent hemoconcentration rather than real renal function impairment.
Time of onset and duration. In contrast to persistent WRF, which is usually associated with hemodynamic derangements and poor prognosis, transient WRF as a result of aggressive decongestive therapy may not be associated with poor outcomes.19
TreatmentMost therapies recommended for AHF lack well-supported evidence.7 In addition, randomized controlled trials in chronic HF and AHF have systematically excluded patients with severe renal dysfunction. For the majority, current therapies include the use of diuretics, inotropic vasoactive agents, and neurohormonal antagonists.
DiureticsLoop diuretics are the pharmacological therapy of choice for the treatment of fluid overload in AHF patients.7 However, their use is largely empirical and is commonly associated with significant deleterious effects, including WRF and a higher risk of worse outcomes.60 Consequently, diuretics have been envisioned as a double-edged sword, with harmful effects in those with renal failure and mild venous congestion, and beneficial effects (renal and prognostic) in patients with severe fluid overload and renal insufficiency.60,62,63 Unfortunately, there are no data in the form of large well-controlled studies aiming to elucidate the optimal diuretic doses for CRS type 1 patients. Indeed, in the DOSE trial, a randomized clinical trial that aimed to investigate the optimal loop diuretic approach in 308 patients with AHF, the authors found that a high- compared with a low-dose strategy, was associated with greater net fluid loss, weight loss, and relief from dyspnea but also with transient WRF.65 Unfortunately, controlled studies evaluating the optimal diuretic strategy in CRS type 1 are still lacking.
UltrafiltrationThe UNLOAD trial81 was a prospective, randomized multicentric trial comparing the effects of early ultrafiltration alone vs intravenous diuretics alone on weight loss, symptoms, and short-term hospitalizations in AHF and volume overload patients with a mean (standard deviation) serum creatinine of 1.5 (0.5) mg/dL. In this study, the authors found no significant renal changes between groups but a superiority of ultrafiltration regarding efficacy endpoints. However, more recently, Bart et al82 assessed the efficacy and safety of ultrafiltration in 188 patients with acute decompensated HF complicated by persistent congestion and WRF. At 96h following enrollment, patients in the ultrafiltration group had a similar effect on weight loss compared with those receiving stepped pharmacologic therapy. However, there was a higher increase in serum creatinine levels in the ultrafiltration group compared with the group treated pharmacologically (0.23 [0.70] mg/dL vs 0.04 [0.53] mg/dL); P = .003); by the same token, the ultrafiltration group had an increased incidence of serious adverse events.82
DopamineClassically, dopamine therapy has been indicated to facilitate diuresis, presumably improving renal blood flow mediated through a modest increase in cardiac output. Among 60 acute decompensated HF patients enrolled in the DAD-HF I trial, WRF occurred more frequently in the high-dose furosemide treatment group than in the group receiving low-dose furosemide combined with low-dose dopamine (30% in high-dose vs 6.7% in low-dose furosemide; P = .042).83 Nevertheless, a recent controlled clinical trial (ROSE)84, which included 360 patients with AHF and renal dysfunction (estimated GFR of 15-60mL/min/1.73 m2), failed to demonstrate the superiority of dopamine therapy on cumulative urine volume and on changes in plasma cystatin C from baseline to 72h.
Renin Angiotensin Aldosterone BlockersThe role of the renin-angiotensin-aldosterone system blockade, with angiotensin-converting enzyme inhibitors or angiotensin receptor blockers in CRS is unclear. Patients with AHF may develop hypotension and/or WRF during initial therapy.7 In patients admitted with significant WRF, current guidelines recommend a reduction or temporary discontinuation of angiotensin-converting enzyme inhibitors or angiotensin receptor blockers until renal function improves.7 Along these lines, evidence supporting the role of aldosterone blockers in CRS is even more scarce.7
Relaxin and Other TherapiesIn the RELAX-AHF trial,84 serelaxin (a synthetic formulation of the hormone relaxin) was associated with a lower incidence of WRF at day 2, lower serum creatinine and plasma cystatin-C values in the first 5 days after enrolment, and a reduction in the risk of 180-day mortality. Conversely, in a large randomized clinical trial of 2033 patients admitted with AHF and renal dysfunction, the adenosine A1 receptor antagonist, rolofylline, failed to demonstrate superiority on the outcome of changes in creatinine and the development of persistent WRF.85
Other therapies, such as vasopressin antagonists, natriuretic peptides, and levosimendan, with potential beneficial effects in CRS type I patients, have either not been rigorously tested or, due to preliminary promising results, they are still under investigation.34
CONCLUSIONWorsening renal function that occurs in AHF is a common finding with a complex and poorly understood pathophysiology. However, the renal function changes observed during HF decompensations should be placed in the right context to accurately differentiate true AKI from WRF due to aggressive decongestion. Thus, a comprehensive assessment should follow these renal function changes in AHF. Future studies are required to obtain further insight into the pathophysiological mechanisms of CRS type I and to search for ways to improve the diagnostic and prognostic accuracy of current methods, as well as to explore effective treatment methods.
FUNDINGThis study was supported by grants from Instituto de Salud Carlos III, Red de Investigación Cardiovascular, Programa 7, (RD12/0042/0010 and RD12/0042/0068) of the Fondo Europeo de Desarrollo Regional.
CONFLICTS OF INTERESTNone declared.