




Danon disease (DD) is caused by mutations in the LAMP2 gene. It is considered a multisystemic disease characterized by hypertrophic cardiomyopathy with pre-excitation and extreme hypertrophy, intellectual disability, myopathy, childhood presentation, and worse prognosis in men. There are scarce data on the clinical characteristics and prognosis of DD.
MethodsWe analyzed the clinical records of patients with DD from 10 Spanish hospitals.
ResultsTwenty-seven patients were included (mean age, 31 ± 19 years; 78% women). Male patients showed a high prevalence of extracardiac manifestations: myopathy (80%), learning disorders (83%), and visual alterations (60%), which were uncommon findings in women (5%, 0%, and 27%, respectively). Although hypertrophic cardiomyopathy was the most common form of heart disease (61%), the mean maximum wall thickness was 15 ± 7 mm and dilated cardiomyopathy was present in 12 patients (10 women). Pre-excitation was found in only 11 patients (49%). Age at presentation was older than 20 years in 16 patients (65%). After a median follow-up of 4 years (interquartile range, 2-9), 4 men (67%) and 9 women (43%) died or required a transplant. Cardiac disease and adverse events occurred later in women (37 ± 9 vs 23 ± 16 and 36 ± 20 vs 20 ± 11 years, respectively).
ConclusionsThe clinical characteristics of DD differ substantially from traditional descriptions: age at presentation of DD is older, the disease is not multisystemic in women, and pre-excitation is infrequent.
Keywords
Danon disease (DD) is a rare X-linked hereditary glycogen storage disease, caused caused by a mutation in the LAMP2 gene, which encodes lysosome-associated protein 2 (OMIM# 309060.0005).1 Thus, unlike other glycogen storage diseases, DD is caused not by a deficit in acid maltase, but by the alteration in the transport protein LAMP-2. The mutated protein causes systemic accumulation of glycogen that can affect organs such as the heart and skeletal muscles. The clinical course of DD described by Danon et al.2 in 1981 features a classic triad of cardiomyopathy, muscle damage, and intellectual disability. DD has a poor prognosis, and the established view is that symptom onset tends to occur during childhood or adolescence.3
The cardiac involvement that typifies DD is hypertrophic cardiomyopathy (HCM), characterized by markedly thickened ventricular walls and pre-excitation on the electrocardiogram (ECG).3,4
DD is a poorly characterized disease, and there is little information in the literature about its clinical manifestation, course, and prognosis. Moreover, information about DD in women is especially scarce.
The present study analyzed the clinical profile and course of patients on the Spanish Danon registry.
METHODSWe conducted a retrospective analysis of clinical data and test results from 27 DD patients treated in 10 Spanish university hospitals. The patients came from a total of 17 families. The Spanish Danon registry is open to all hospitals wishing to participate and is coordinated from the Puerta de Hierro University Hospital in Majadahonda, Madrid. The patient inclusion criterion is the presence of a genetically confirmed pathogenic mutation in LAMP2.
The pathogenicity of the detected genetic variants (Table 1) was cataloged according to the criteria published by the American College of Medical Genetics and Genomics5: previous description, presence on a publicly available general population database (Exome Aggregation Consortium, Exome Variant Server, and ClinVar) or pathogenicity database (Human Genome Mutation Database), production of a compatible phenotype, and family information.
Frequency and Sex Distribution of Pathogenic LAMP2 Genetic Variants
| Genome position | Protein level (NP_002285.1) | MAFExAC | EVS MAF (%) (EA/AA/all) | ClinVar | HGMD | no. | M (n=6) | F (n=21) | Phenotype |
|---|---|---|---|---|---|---|---|---|---|
| g.119590551C>T | p.Trp46* | NI | NI | NI | CM132566Pathogenic | 6 | 3 | 3 | HCMMyopathy |
| g.119576454C>T | p.Val310Ile | NI | NI | rs104894858Pathogenic | CM057189Pathogenic | 2 | 0 | 2 | HCM |
| g.119580245_119580246insT | p.His260GInfs*14 | NI | NI | NI | NI | 3 | 0 | 3 | HCM |
| ChrX:119573044-119636423del [ChrX:119635816-119635989]ins | NI | NI | NI | NI | 2 | 0 | 2 | HCM | |
| g.119580246G>A | p.His260Tyr | 2/87.696(0.0022) | NI | rs778577575Uncertain significance | NI | 2 | 1 | 1 | HCMPreexcitation |
| g.119575626delC | p.Val352Phefs*6 | NI | NI | NI | NI | 2 | 0 | 2 | DCM |
| g.119589350_119589351insT | p.Ile88Asnfs*25 | NI | NI | NI | NI | 1 | 0 | 1 | DCM |
| g.119582963delG | p.Leu140Phefs*8 | NI | NI | NI | NI | 1 | 0 | 1 | HCMPreexcitation |
| g.119590506delA | p.Tyr61* | NI | NI | NI | NI | 2 | 1 | 1 | DCM |
| g.119576505G>A | p.Arg293* | NI | NI | rs727503118Pathogenic | HM070116Pathogenic | 2 | 0 | 2 | DCMPreexcitation |
| g.119582962delA | p.Leu141Trpfs*7 | NI | NI | NI | NI | 1 | 0 | 1 | HCM |
| g.119581726delC | p.Leu239Cysfs*3 | NI | NI | NI | NI | 1 | 0 | 1 | DCM |
| g.119576454C>A | p.Val310Phe | NI | NI | NI | NI | 1 | 0 | 1 | HCM |
| g.119575602_119575603insT | p.Gly360Argfs*13 | NI | NI | NI | NI | 1 | 1 | 0 | HCM |
AA, African American; ClinVar, Clinical Variant; DCM, dilated cardiomyopathy; EA, European American; ExAC, Exome Aggregation Consortium; EVS, Exome Variant Server; F, female patients; HCM, hypertrophic cardiomyopathy; HGMD, Human Genome Mutation Database; M, male patients; MAF, Minor allele frequency; NI, not identified.
Patient information was collected with a standardized and uniform data form, completed after a thorough review of printed and digital clinical records.
Statistical AnalysisNormally distributed continuous variables are expressed as mean±standard deviation and nonnormally distributed continuous variables as median (range). Categorical variables are expressed as percentages. For between-sex comparisons, 2-sided Student t tests for independent samples were used for normally distributed continuous variables, nonparametric tests for nonnormally distributed continuous variables, and the Fisher exact test for binary variables. Differences were considered statistically significant at P <.05. The statistical analysis was conducted with SPSS version 20 (IBM Corp.; Amronk, New York, United States).
RESULTSData were analyzed from a cohort of 27 patients with pathogenic mutations in the LAMP2 gene. Of the patients, only 6 were men (22%), and 17 patients (63%) were the first in their families to be diagnosed with DD (index cases). Just 5 patients (4 women) were carriers of missense genetic variants (Table 1). The pathogenicity of the variant g.119580246G>A was attested by its DD-compatible phenotype and affected family members.
Of the study population, 21 patients showed signs of disease-related damage at the time of DD diagnosis, 2 developed symptoms during follow-up, and 4 (3 women and 1 man) were carriers showing no signs of damage during the follow-up period. In 2 individuals, the diagnosis was made during postmortem examination (molecular autopsy).
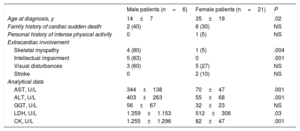
Mean age at diagnosis was 31±19 years. Male patients tended to be diagnosed at a younger age (14±7 years, vs 35±19 years for women), and this difference was statistically significant (P=.02) (Table 2).
Extracardiac Involvement in Danon-disease Patients by Sex: Clinical Characteristics and Analytical Findings
| Male patients (n=6) | Female patients (n=21) | P | |
|---|---|---|---|
| Age at diagnosis, y | 14±7 | 35±19 | .02 |
| Family history of cardiac sudden death | 2 (40) | 6 (30) | NS |
| Personal history of intense physical activity | 0 | 1 (5) | NS |
| Extracardiac involvement | |||
| Skeletal myopathy | 4 (80) | 1 (5) | .004 |
| Intellectual impairment | 5 (83) | 0 | .001 |
| Visual disturbances | 3 (60) | 5 (27) | NS |
| Stroke | 0 | 2 (10) | NS |
| Analytical data | |||
| AST, U/L | 344±138 | 70±47 | .001 |
| ALT, U/L | 403±263 | 55±68 | .001 |
| GGT, U/L | 56±67 | 32±23 | NS |
| LDH, U/L | 1.359±1.153 | 512±306 | .03 |
| CK, U/L | 1.255±1.296 | 82±47 | .001 |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; GGT, gamma-glutamyl transpeptidase; LDH, lactate dehydrogenase; NS, nonsignificant.
Data are expressed as No. (%) or mean±standard deviation.
A family history of sudden cardiac death was recorded in 8 patients (29% of the cohort).
Multisystem InvolvementWomen showed only sporadic extracardiac involvement, affecting skeletal muscle, eyesight, liver, or intellectual capacity (Table 2 and Figure 1). For example, only 5% of female patients had skeletal myopathy, compared with 80% of male patients. These rates correlated with significantly higher serum creatine kinase in male than in female patients (1255±1296 U/L vs 82±47 U/L; P <.05). The 5 patients with learning disorders were all male (83% of the male study population). Intellectual disability in these patients was mild to moderate, with all of them maintaining full autonomy in day-to-day activities.

Liver enzyme elevation was also more frequent in male patients, reflected in higher mean levels of AST (aspartate aminotransferase) and ALT (alanine aminotransferase) (Table 2).
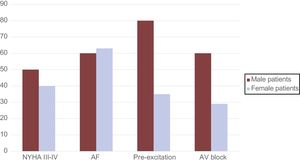
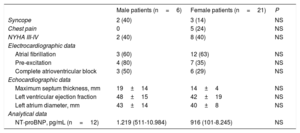
Cardiac DamageThe cardiac symptoms shown by the DD patients were syncope (22% of patients), chest pain (22%), and heart failure (New York Heart Association functional class III-IV; 43%) (Table 3).
Cardiac Involvement in Danon-disease Patients by Sex
| Male patients (n=6) | Female patients (n=21) | P | |
|---|---|---|---|
| Syncope | 2 (40) | 3 (14) | NS |
| Chest pain | 0 | 5 (24) | NS |
| NYHA III-IV | 2 (40) | 8 (40) | NS |
| Electrocardiographic data | |||
| Atrial fibrillation | 3 (60) | 12 (63) | NS |
| Pre-excitation | 4 (80) | 7 (35) | NS |
| Complete atrioventricular block | 3 (50) | 6 (29) | NS |
| Echocardiographic data | |||
| Maximum septum thickness, mm | 19±14 | 14±4 | NS |
| Left ventricular ejection fraction | 48±15 | 42±19 | NS |
| Left atrium diameter, mm | 43±14 | 40±8 | NS |
| Analytical data | |||
| NT-proBNP, pg/mL (n=12) | 1.219 (511-10.984) | 916 (101-8.245) | NS |
NS, nonsignificant; NT-proBNP, N-terminal pro-brain natriuretic peptide; NYHA, New York Heart Association.
Data are expressed as No. (%) or mean±standard deviation.
A systematic analysis of available DD-patient ECGs in the registry revealed a preexcitation pattern in only 11 patients (48%), 4 of whom were male (Figure 2). Atrial fibrillation (Figure 3) was very common, affecting half of the male and female patients in the registry. In contrast, whereas 4 women developed sustained ventricular tachycardia during follow-up (with the condition deteriorating to ventricular fibrillation in 1 patient), none of the male patients had these arrhythmias.


Electrocardiographic findings in 2 patients with Danon disease. A, ECG in a 14-year-old male patient with severe ventricular hypertrophy and preexcitation. The ECG was recorded at half voltage. B, ECG of a 23-year-old male patient, with no preexcitation and no signs of ventricular hypertrophy. ECG, electrocardiogram.
In the series, 11 patients (41%; 8 female and 3 male) received an implantable cardioverter defibrillator (ICD). An ICD was indicated for secondary prevention in only 1 patient, a woman with a clear phenotype (HCM and pre-excitation) who developed sustained ventricular tachycardia. ICD implantation for primary prevention was indicated in patients with ventricular dysfunction or a family history of sudden cardiac death. During follow-up, appropriate ICD discharges were documented in 1 male patient, and another was treated for sustained ventricular tachycardia that developed in the context of inotropic medication with levosimendan. No other patient with an ICD developed ventricular arrhythmias.
Cardiac conduction disorders were also common. Overall, 50% of male patients (n=3) and 29% of women (n=6) experienced complete atrioventricular block, with no statistically significant differences between subgroups.
All patients tested (n=12) showed a generalized elevation of N-terminal pro-brain natriuretic peptide, with a tendency toward higher concentrations in male patients than in women: 1219 (511-10 984) pg/mL vs 916 (101-8245) pg/mL (P> .05).
Echocardiography data revealed a mean left ventricular thickness of 15±7mm and a slightly depressed mean left ventricular ejection fraction (LVEF) (43±18%). Interestingly, in 12 patients in the registry (44%) the cardiomyopathy at the time of diagnosis was dilated cardiomyopathy (DCM), with a mean LVEF of 27±8%. Women showed a tendency toward thinner ventricular walls (14±4mm vs 19±14mm; P=.21) and a lower LVEF (42±19% vs 48±15%; P=.49). At the time of diagnosis, 48% of female patients had DCM vs 33% of male patients (P=.54). The mutation type was not associated with statistically significant differences in the recorded characteristics. Nevertheless, the phenotype of the 5 missense-variant carriers fell broadly within the pattern of HCM with preserved LVEF. Moreover, these patients showed little multisystem involvement measured by clinical or analytical parameters (Table 4).
Extracardiac and Cardiac Involvement in Danon-disease Patients by Mutation Type
| Missense (n=5) | Not missense (n=22) | P | |
|---|---|---|---|
| Age at diagnosis, y | 30±16 | 31±4 | NS |
| Extracardiac involvement | |||
| Skeletal myopathy | 0 | 5 (23) | NS |
| Intellectual impairment | 0 | 5 (23) | NS |
| Visual disturbances | 1 (20) | 7 (32) | NS |
| Analytical data | |||
| AST, U/L | 84±17 | 129±33 | NS |
| ALT, U/L | 41±12 | 135±46 | NS |
| GGT, U/L | 18±3 | 40±9 | NS |
| LDH, U/L | 923 | 710±184 | NS |
| CK, U/L | 114±34 | 352±185 | NS |
| Cardiac involvement | |||
| Maximal septal thickness, mm | 17±4 | 15±2 | NS |
| Left ventricular ejection fraction | 58±5 | 40±4 | NS |
| Preexcitation | 1 (20) | 10 (46) | NS |
| NYHA III-IV | 1 (20) | 9 (41) | NS |
ALT, alanine aminotransferase; AST, aspartate aminotransferase; CK, creatine kinase; GGT, gamma-glutamyl transpeptidase; LDH, lactate dehydrogenase; NS, nonsignificant; NYHA, New York Heart Association.
Data are expressed as No. (%) or mean±standard deviation.
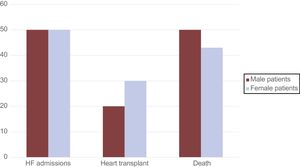
Excluding patients diagnosed at postmortem, median follow-up was 4 years [interquartile range, 2-9]. During this period, 48% of these DD patients were admitted for heart failure (50% of male patients and 47% of female patients) (Table 5). The mean age at the first heart failure episode was 23±16 years in male patients and 37±9 years in women (P=.09).
Events and Age at Onset in Danon-disease Patients by Sex
| Male patients (n=6) | Female patients (n=21) | P | |
|---|---|---|---|
| Admission for heart failure | 2 (50) | 10 (47) | NS |
| Age at first heart-failure episode, y | 23±16 | 37±9 | NS |
| Heart transplant | 1 (20) | 6 (30) | NS |
| Age at heart transplant, y (n=7) | 14 | 32±9 | NS |
| Death | 3 (50) | 3 (14) | NS |
| Age at death, y (n=6) | 23±13 | 40±14 | NS |
| Death or heart transplant | 4 (57) | 9 (43) | NS |
| Age at death or heart transplant (n=13) | 20±11 | 36±20 | NS |
NS, nonsignificant.
Data are expressed as No. (%) or mean±standard deviation.
In the cohort, 6 deaths were recorded (22% of the total population) (Figure 4). There were 3 male deaths (50%): 1 due to heart failure and 2 due to sudden cardiac death. Of the female deaths, 2 were due to advanced heart failure and 1 to sudden cardiac death. Heart transplants were performed in 7 patients, including 1 male patient. This patient received the transplant at the age of 14 years, whereas the mean age of female heart transplant recipients was 32±9 years (range, 18-43 years).

In line with the time of heart failure onset, the mean age of heart transplant or death was higher among female than among male patients, although the difference did not reach statistical significance (36±20 years vs 20±11 years; P=.14) (Figure 4).
DISCUSSIONThis study presents the patient characteristics and clinical course for one of the largest cohorts of DD patients described to date. The analysis of the DD patients in this cohort reveals a clinical profile substantially different from that historically described for this disease. Our data show that DD does not usually manifest with multisystem involvement in women, that pre-excitation on the ECG is an infrequent disease feature, and that the age of disease onset is later than described previously. Moreover, our data show that although women with DD have an unfavorable prognosis, they experience adverse events at a substantially more advanced age than their male counterparts.
DD is a rare X-linked inherited disease caused by a mutation in the LAMP2 gene. Pathogenic LAMP2 mutations result in total or partial loss of the enzyme LAMP-2, and the loss of LAMP-2 activity leads to lysosomal glycogen accumulation, which is the histopathological basis of DD. To date, more than 100 DD-causing LAMP2 mutations have been reported.6
DD is regarded as a multisystem disease characterized by a classic triad of intellectual disability, skeletal myopathy, and HCM featuring severe ventricular hypertrophy and pre-excitation on the ECG.3,4 Less frequently, DD patients can also develop retinopathy and lung or liver disease,2 although this was very rare in our patients. As observed in our series, the onset of this X-linked disease is generally earlier and the clinical presentation worse in male patients, with disease symptoms appearing during childhood or adolescence.3 In contrast, the heterozygous state of women with DD results in infrequent symptom onset before adulthood.7
The exact prevalence of DD is unknown, but recent reports suggest that it is higher than previous estimates and could be as high as 1% to 6% of patients with HCM of unknown etiology.4,8 The prevalence could reach 17% if we limit consideration to patients in whom HCM is accompanied by other features, such as elevated creatine kinase or preexcitation on the ECG.4
Skeletal myopathy in DD patients manifests as weakness, exercise intolerance, and elevated serum creatine kinase. However, in our population, this triad was found only in male patients, and only 1 of the 21 women in the registry complained of muscle pain.
Learning difficulties were found in only 19% of DD patients, a much lower rate than established in the literature (70%-100% of male patients and up to 47% of women).6 In line with our findings, a recent detailed psychological and cognitive evaluation of DD patients found that cognitive and psychiatric changes are uncommon.9 On the other hand, the same study reported frequent mood disorders and anxiety, affecting as many as 70% of DD patients independently of sex.9
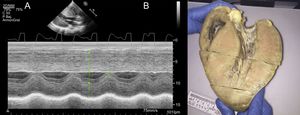
The most frequently reported cardiac involvement in DD is HCM with preserved LEVF, and DD is therefore considered a phenocopy of HCM.10 Moreover, LAMP-2 cardiomyopathy tends to have a worse prognosis than HCM caused by sarcomere mutations.8,11 Cardiac hypertrophy in DD patients can be extreme; an example is provided by the heart of a 14-year-old male patient in our cohort who underwent heart transplant (Figure 5). Indeed, the largest heart reported in the literature to date was from a 14-year-old American male DD patient, whose autopsy revealed a wall thickness of 65mm and a heart mass of 1425g.3
 and macroscopic image (B) of the heart of a 14-year-old male patient with Danon disease featuring severe left ventricular hypertrophy (interventricular septum, 43mm). Macroscopic image courtesy of Dr. Elena Ruiz, Hospital de la Paz, Madrid.")
Although HCM is the most common form of cardiac involvement in DD, it is important to note that a subgroup of patients develops DCM. This subgroup is predominantly composed of women and in our cohort included 41% of women with any type of cardiomyopathy. It is therefore a mistake to exclude a diagnosis of DD in patients with DCM; indeed DD should be suspected in patients with HCM that progresses to DCM.9,12 It is also recommended not to restrict LAMP2 tests to patients with HCM, but to include this gene in genetic studies of patients with DCM.10
Patients with DD frequently develop conduction abnormalities and arrhythmias. Pre-excitation on ECG has been reported in 68% of male DD patients and 27% of women with this disease7 and is an important indicator of suspected DD.10 However, in our series, pre-excitation was less frequent than in previous reports, affecting just 41% of patients. This low rate could make it harder to reach a firm diagnosis in some patients.
High rates of ventricular arrhythmias in DD patients, especially male patients, have been reported previously in several smaller series than ours. In our series, there were 3 instances of sudden cardiac death, in 2 male patients and 1 woman, none of whom had an ICD; 4 other patients (all women) had sustained ventricular tachycardia during follow-up. The combined rate of severe arrhythmia and sudden cardiac death in this cohort is high (26% of patients) and is higher than a recently reported combined ventricular arrhythmia/sudden cardiac death rate of around 14%.13
The data from our registry confirm that DD has a poor prognosis in both sexes and that without heart transplant male patients do not reach the age of 25 years. Likewise, it is noteworthy that diverse incidences of ICD discharge have been reported that did not achieve termination of ventricular arrhythmias in DD patients.3
It is therefore vitally important to diagnose DD early, based on a high level of vigilance. Early diagnosis is necessary to allow physicians to devise treatment strategies and care plans that can anticipate complications in these patients.
There are currently no specific guidelines on the treatment of DD, and there have been no clinical trials in this area. Indeed, the indications for an ICD or heart transplant in DD are similar to those established for HCM and DCM. Thus, for DD patients with an HCM phenotype, the indication is based on classic risk factors for sudden cardiac death and HCM. In contrast, for a DD patient with DCM, the indication for an ICD is based on LVEF and functional class. A set of professional recommendations was recently published on DD follow-up and treatment.14 This report placed strong emphasis on the need for a multidisciplinary approach to DD, stressing the need for the medical team to include cardiologists, neurologists, ophthalmologists, psychiatrists, and clinical geneticists.
LimitationsAlthough our DD patient series is one of the largest reported to date, it is important to acknowledge that the number of patients is small. In addition, the data were compiled retrospectively by reviewing patient medical histories, which can introduce bias in information, survival, or selection. Related to this, parameters such as intellectual disability were not evaluated using specific study protocols. It is also important to note the small number of male patients and the possible implications of this for the statistical analysis. Moreover, the multicenter nature of the registry may have skewed the selection to underrepresent patients with a milder clinical manifestation.
CONCLUSIONSDD has a poor prognosis linked to the associated cardiac involvement. Cardiac involvement occurs in both sexes, but normally appears later in women.
The absence of the classic triad does not justify ruling out a diagnosis of DD, especially in women, who can show exclusively cardiac involvement. Moreover, symptoms regarded as defining characteristics, such as pre-excitation on the ECG, can be present in only a minority of patients with DD.
A deeper knowledge of the clinical characteristics and the pathophysiological mechanisms of DD would assist the diagnosis of this disorder. Appropriate treatment of DD requires the definition of a strategy that includes early evaluation for heart transplant.
FUNDINGThis study received funding from the Carlos III Institute of Health (PI14/01477 and the La Fe Biobank PT17/0015/0043), which is supported by the European Regional Development Fund (ERDF) “A way to make Europe”.
CONFLICTS OF INTERESTNone declared.
- –
DD is a rare X-linked inherited disease caused by mutations in the LAMP2 gene. It is regarded as a multisystem disease characterized by HCM with pre-excitation and severe hypertrophy, intellectual disability, skeletal myopathy, childhood onset, and a worse prognosis in male patients.
- –
DD is a poorly characterized disease and there are few published series. There is therefore little available information on the treatment, progression, and prognosis of this disease. Moreover, data are especially scarce in women with DD.
- –
The clinical characteristics of DD patients differ substantially from the traditional picture of this disease. According to our data, DD does not manifest as a multisystem disease in women, who have a later disease onset. Pre-excitation on the ECG was found in a minority of patients. Cardiac involvement is the main prognostic factor, and males not receiving a heart transplant do not reach the age of 25 years. Although prognosis is also poor in women, adverse events appear at a later age. Our data indicate that DD could be considered to phenocopy both HCM and DMC, since in a sizable proportion of patients (especially women) cardiac involvement manifests directly as DCM.
We thank Dr Herminio Morillas for patient referrals and Dr Elena Ruiz for providing the macroscopic image in Figure 5.