Cardiac amyloidosis (CA) is produced by amyloid fiber deposition in the myocardium. The most frequent forms are those caused by light chains (AL) and transthyretin (ATTR). Our objective was to describe the diagnosis, treatment and outcomes of CA in a specialized Spanish center.
MethodsWe included all patients diagnosed with CA in Hospital Universitario Puerta de Hierro Majadahonda from May 2008 to September 2018. We analyzed their clinical characteristics, outcomes, and survival.
ResultsWe included 180 patients with CA, of whom 64 (36%) had AL (50% men; mean age, 65±11 years) and 116 had ATTR (72% men; mean age 79±11 years; 18 with hereditary ATTR). The most common presentation was heart failure in both groups (81% in AL and 45% in ATTR, P <.01). Other forms of presentation in ATTR patients were atrial arrhythmias (16%), conduction disorders (6%), and incidental finding (6%); 70 patients (40%), had a previous alternative cardiac diagnosis. Diagnosis was noninvasive in 75% of ATTR patients. Diagnostic delay was higher in ATTR (2.8±4.3 vs 0.6±0.7 years, P <.001), but mortality was greater in AL patients (48% vs 32%, P=.028). Independent predictors of mortality were AL subtype (HR, 6.16; 95%CI, 1.56-24.30; P=.01), female sex (HR, 2.35; 95%CI, 1.24-4.46; P=.01), and NYHA functional class III-IV (HR, 2.07; 95%CI, 1.11-3.89; P=.02).
ConclusionsCA is a clinical challenge, with wide variability in its presentation depending on the subtype, leading to diagnostic delay and high mortality. Improvements are needed in the early diagnosis and treatment of these patients.
Keywords
The term cardiac amyloidosis (CA) refers to a heart condition associated with amyloid fibril deposition.1 In Spain, the most frequent CA subtypes are amyloid light-chain amyloidosis (AL), caused by the deposition of light chains from plasma cell dyscrasia, and transthyretin (TTR) amyloidosis (ATTR), caused by the deposition of TTR. The latter has 2 forms: the hereditary variant (hATTR) and the nonhereditary or wildtype form (ATTRwt), previously called senile amyloidosis.1,2
Classically, the AL form was believed to be the most prevalent amyloidosis subtype in developed countries.3 However, due to population aging and the use of noninvasive diagnostic techniques,4 ATTR is now considered the most frequent form of CA.5
CA is thus far an underdiagnosed condition with a major diagnostic delay.6 This delay is partly due to the traditional consideration of CA as an untreatable disease, as well as the complexity of its diagnosis, which includes the need for histological evidence.2,5 Nonetheless, CA can now be diagnosed without the need for histological confirmation in a considerable number of patients with CA and major advances have been made in both diagnostic techniques and treatment.7–13
The last decade has seen the development of new therapies with promising results for both ATTR and AL.7–10 Some of these drugs have shown very satisfactory results in clinical trials.10,11 Identification of those patients who will benefit from the new treatments is essential, particularly at early stages, when the therapies are more effective.
The experience with CA patient diagnosis and treatment is thus far slight and confined to a few centers. With the increase in the number of patients diagnosed and the development of new therapies, widespread dissemination is required of the characteristics of the presentation, prognosis, and clinical course and treatment of CA patients.
The objective of the present study was to analyze the characteristics of CA patients diagnosed and followed up in a Spanish referral center during a 10-year period.
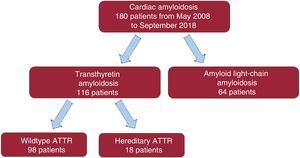
METHODSThis descriptive, observational, and retrospective study examined all consecutive patients diagnosed with CA in Hospital Universitario Puerta de Hierro Majadahonda from May 2008 to September 2018 (figure 1). The study was approved by the ethics committee of the center.

Invasively and noninvasively diagnosed patients were included. The invasive diagnosis was based on histological documentation of amyloid in endomyocardial biopsy (EMB) or extracardiac biopsy when echocardiography or magnetic resonance revealed the presence of left ventricular hypertrophy (LVH) (≥ 12mm), after the exclusion of other causes of LVH.12 To differentiate AL and ATTR, immunohistochemistry was performed using monoclonal antibodies against AA, TTR, and light chains (DAKO, Denmark). In doubtful cases or those with positive staining with multiple antibodies, mass spectrometry was performed for the definitive diagnosis of the amyloid subtype.
ATTR was noninvasively diagnosed according to published criteria13 and involved the presence of data compatible with CA from cardiac imaging techniques and a Perugini score of 2 or 3 on 99mTC-DPD scintigraphy,4 after ruling out AL using analysis of serum light chains (Freelite, The Binding Site Group, United Kingdom) and blood and urine immunofixation. A genetic study was performed in all ATTR patients involving complete sequencing of the TTR gene.
The hematological results indicated monoclonal gammopathy of undetermined significance and plasma cell dyscrasia, including multiple myeloma, as well as the need for bone marrow biopsy according to current criteria14 after hematological assessment
The study did not include patients with mutations in the TTR gene or those with AL without cardiac involvement.
VariablesThe information was obtained through medical record review. Data were obtained on the first evaluation and follow-up.
Data on the final patient status were obtained from the hospital and primary care medical records. Total mortality was defined as death from any cause during follow-up.
Statistical analysisVariable normality was studied with the Shapiro-Wilk test. Normal continuous variables are expressed as mean ± standard deviation. Categorical variables are expressed as percentages. To compare continuous variables, the 2-tailed Student t test was used for independent samples; the chi-square test or Fisher test was used for categorical data. The Kaplan-Meier curve was used to calculate survival. P < .05 was considered statistically significant.
An analysis of the time to death or heart transplantation was performed in patients with AL and with ATTR. In addition, a multivariate analysis of survival predictors was performed using a model adjusted for age, sex, CA type, and variables with P < .10 in the univariable model (): heart failure (HF) at diagnosis, New York Association (NYHA) functional class III-IV at diagnosis, N-terminal pro-B-type natriuretic peptide (NT-proBNP) > 3000 ng/dL, glomerular filtration rate (continuous and < 45mL/min/1.73 m2), and left ventricular ejection fraction (LVEF) < 50%.
STATA version 12.0 (StataCorp LP, United States) was used for the statistical analysis.
RESULTSIn total, 180 patients with CA were included (64.1% men; mean age at diagnosis, 74.3 ± 12.7 years); 64 were diagnosed were AL (35.6%; 50% men; age, 64.9 ± 11.1 years) and 116 with ATTR (64.4%; 71.6% men; age, 79.4 ± 10.5 years). Of the latter, 98 (77% men; age, 80.8 ± 8.5 years) had ATTRwt and 18 (44% men; age, 68.9 ± 14.6 years) had hATTR (figure 1). The patients’ baseline characteristics are detailed in table 1.
Clinical profile at diagnosis and previous diagnosis
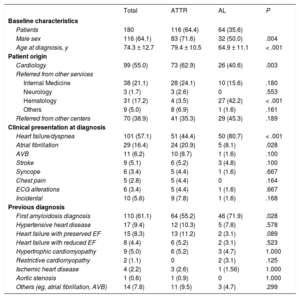
| Total | ATTR | AL | P | |
|---|---|---|---|---|
| Baseline characteristics | ||||
| Patients | 180 | 116 (64.4) | 64 (35.6) | |
| Male sex | 116 (64.1) | 83 (71.6) | 32 (50.0) | .004 |
| Age at diagnosis, y | 74.3 ± 12.7 | 79.4 ± 10.5 | 64.9 ± 11.1 | < .001 |
| Patient origin | ||||
| Cardiology | 99 (55.0) | 73 (62.9) | 26 (40.6) | .003 |
| Referred from other services | ||||
| Internal Medicine | 38 (21.1) | 28 (24.1) | 10 (15.6) | .180 |
| Neurology | 3 (1.7) | 3 (2.6) | 0 | .553 |
| Hematology | 31 (17.2) | 4 (3.5) | 27 (42.2) | < .001 |
| Others | 9 (5.0) | 8 (6.9) | 1 (1.6) | .161 |
| Referred from other centers | 70 (38.9) | 41 (35.3) | 29 (45.3) | .189 |
| Clinical presentation at diagnosis | ||||
| Heart failure/dyspnea | 101 (57.1) | 51 (44.4) | 50 (80.7) | < .001 |
| Atrial fibrillation | 29 (16.4) | 24 (20.9) | 5 (8.1) | .028 |
| AVB | 11 (6.2) | 10 (8.7) | 1 (1.6) | .100 |
| Stroke | 9 (5.1) | 6 (5.2) | 3 (4.8) | .100 |
| Syncope | 6 (3.4) | 5 (4.4) | 1 (1.6) | .667 |
| Chest pain | 5 (2.8) | 5 (4.4) | 0 | .164 |
| ECG alterations | 6 (3.4) | 5 (4.4) | 1 (1.6) | .667 |
| Incidental | 10 (5.6) | 9 (7.8) | 1 (1.6) | .168 |
| Previous diagnosis | ||||
| First amyloidosis diagnosis | 110 (61.1) | 64 (55.2) | 46 (71.9) | .028 |
| Hypertensive heart disease | 17 (9.4) | 12 (10.3) | 5 (7.8) | .578 |
| Heart failure with preserved EF | 15 (8.3) | 13 (11.2) | 2 (3.1) | .089 |
| Heart failure with reduced EF | 8 (4.4) | 6 (5.2) | 2 (3.1) | .523 |
| Hypertrophic cardiomyopathy | 9 (5.0) | 6 (5.2) | 3 (4.7) | 1.000 |
| Restrictive cardiomyopathy | 2 (1.1) | 0 | 2 (3.1) | .125 |
| Ischemic heart disease | 4 (2.2) | 3 (2.6) | 1 (1.56) | 1.000 |
| Aortic stenosis | 1 (0.6) | 1 (0.9) | 0 | 1.000 |
| Others (eg, atrial fibrillation, AVB) | 14 (7.8) | 11 (9.5) | 3 (4.7) | .299 |
AL, amyloid light-chain amyloidosis; ATTR, transthyretin amyloidosis; AVB, atrioventricular block; ECG, electrocardiography; EF, ejection fraction.
Values represent No. (%) or mean ± standard deviation.
Between May 2008 and September 2018, 223 biopsies were performed: 61 EMBs (34%), 71 of the bone marrow (41%), and 91 of other organs (51%). The EMBs were mainly performed in the right ventricle (56; 91.8%); only 5 (8.2%) were obtained from the left ventricle. The complication rate was low considering the invasiveness of the technique: only 2 complications (3.3%) were recorded, both nonfatal. The complications occurred after EMB of the right ventricle and comprised 1 intraprocedural ventricular arrhythmia and 1 mild effusion with tamponade due to anticoagulant therapy. The EMB yield was 98.4% and a diagnosis was made in 56% of AL patients (n = 36). In 1 patient, left ventricular EMB was performed because no amyloid was found in the right ventricular sample. Regarding extracardiac biopsies, 16 were obtained from the salivary gland, 20 from the abdominal fat, 8 from the gastrointestinal tract, 6 from the kidneys, and 1 from the peripheral nerves. The yields varied but tended to be low, with the exception of the renal and peripheral nerve biopsies, and depended on the organ targeted (19%, 40%, 60%, 80%, and 100%, respectively). In the bone marrow biopsies performed in patients suspected to have AL, a plasma cell component exceeding 10% was found in 56% of patients (36 patients) and amyloid in 81% (52 patients).
The definitive diagnosis of amyloidosis subtype was obtained using mass spectrometry in 8 patients (4.7%). The main reason for the use of this technique was double immunohistochemical staining for both TTR and one of the light chains. The proteomic results identified 2 of the patients as having AL and 6 as having ATTR, in line with the initial clinical suspicion in all patients except 1.
ATTR-CA was noninvasively diagnosed in 87 patients (75%). EMB was only required in 25 of them (22%). Four patients were diagnosed due to cardiac involvement and a positive extracardiac biopsy (3 of the abdominal fat and 1 of the gastrointestinal tract).
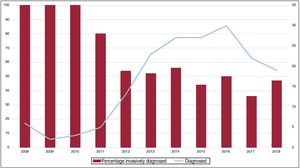
Although the percentage of patients with a noninvasive diagnosis in the entire series has increased in recent years, a considerable number are still diagnosed through biopsy (figure 2).
 and total number of patients with CA diagnosed per year.")
The TTR gene was analyzed in all individuals diagnosed with ATTR-CA and 18 patients (16%) were found to have the following mutations: 6 had Val50Met, 6 had Val142Ile, 3 had Glu109Lys, 1 had Glu109Gln, 1 had Thr80Ala, and 1 had Ala65Thr. Of the patients diagnosed with hATTR, 14 (78%) were the first members of their families diagnosed. Among the 114 patients with ATTR older than 60 years, a mutation was found in 12% (14 patients). These results enabled the genetic study of 57 first-degree relatives belonging to 16 families, and 20 other individuals (35%) were found to have mutations in the TTR gene.
99mTc-DPD scintigraphy was performed in 165 patients (92%). The majority of these patients had ATTR (112; 68%); most of these patients (91%) had a Perugini score of 3. Of the 53 patients with AL who underwent 99mTc-DPD scintigraphy, 32% (n = 17) had some degree of uptake: 7 patients had grade 3 uptake, 6 had grade 2, and 4 had grade 1. Of the 13 patients with AL and grade 2 or 3 uptake, 7 had uptake in the left ventricle alone, 5 in both ventricles, and 1 in the right ventricle alone.
The time from symptom onset to definitive diagnosis was significantly longer in the ATTR group, with a mean of 2.8 ± 4.3 vs 0.6 ± 0.7 years for AL (P < .001); 70 patients (40%) had already been diagnosed with a different cardiac condition (table 1). The previous diagnosis was obtained in 45% and 28% of patients with ATTR and AL, respectively. The most frequent diagnosis in both subtypes together was hypertensive heart disease (17 patients; 9.4%), followed by HF with preserved LVEF, which was particularly common in patients with ATTR (12; 10%).
Patient origin and clinical presentationMost of the patients diagnosed with CA were derived from cardiology (99; 55%), followed by hematology (31 patients; 17%), from where 42% of the AL patients originated, as well as 4% of the ATTR. Of the patients with ATTR, 41% met the criteria for monoclonal gammopathy of undetermined significance.
Although there is particular interest in this disease in our center and we have long had a systematic disease diagnosis system in place for patients admitted with HF and increased ventricular thickness, more than one third of patients (70; 39%) were derived from other centers (table 1). Proportionally, referrals were more frequent for patients with AL (45%) than for those with ATTR (35%).
Regarding the clinical presentation, HF/dyspnea was the most common and the initial presentation in 81% of the AL patients vs only 44% of the ATTR patients (P < .001). In contrast to AL patients, the clinical presentation prompting the diagnosis was much more variable in ATTR patients and ranged from atrial fibrillation (21% in ATTR vs 8% in AL; P = .028) and conduction disorders (6%) to electrocardiographic abnormalities (3%) or were obtained during the etiological study of stroke (5%) or as an incidental finding after scintigraphy for rheumatological or oncological reasons (6%).
CA was diagnosed during admission in 53% of patients (69% for AL, 37% for hATTR, and 45% for ATTRwt).
Cardiac involvementUp to one third of patients (61; 34%) were in NYHA III-IV functional class at diagnosis. More than half of the patients (106; 59%) had already required admission due to HF, particularly the AL subgroup (70% vs 53%; P = .021). However, the ATTR subgroup contained a higher percentage of patients with a history of atrial fibrillation (50% vs 16%; P < .001) and a higher rate of pacemakers at the first assessment (21% vs 5%; P = .004). Three patients (2%) had an implantable cardioverter-defibrillator at the first evaluation.
Regarding biomarkers, the mean NT-proBNP level was 7873 ± 11 312 pg/mL. It was significantly higher in AL than in ATTR (13 338 ± 16 064 vs 4805 ± 5492 pg/mL; P < .001) and in patients diagnosed during admission vs those diagnosed without being admitted (10 987 ± 13 704 vs 4308 ± 6068 pg/mL; P < .001). There were no significant differences in troponin I levels between AL and ATTR (0.18 ± 0.56 vs 0.20 ± 0.21μg/L; P = .843). There were also no differences in glomerular filtration rates, although patients with AL more frequently had dipstick-positive proteinuria (table 2).
Diagnosis and clinical characteristics of patients with cardiac amyloidosis according to subtype
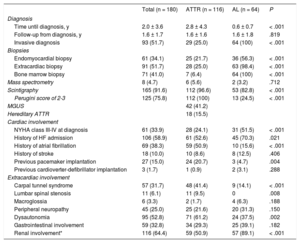
| Total (n = 180) | ATTR (n = 116) | AL (n = 64) | P | |
|---|---|---|---|---|
| Diagnosis | ||||
| Time until diagnosis, y | 2.0 ± 3.6 | 2.8 ± 4.3 | 0.6 ± 0.7 | < .001 |
| Follow-up from diagnosis, y | 1.6 ± 1.7 | 1.6 ± 1.6 | 1.6 ± 1.8 | .819 |
| Invasive diagnosis | 93 (51.7) | 29 (25.0) | 64 (100) | < .001 |
| Biopsies | ||||
| Endomyocardial biopsy | 61 (34.1) | 25 (21.7) | 36 (56.3) | < .001 |
| Extracardiac biopsy | 91 (51.7) | 28 (25.0) | 63 (98.4) | < .001 |
| Bone marrow biopsy | 71 (41.0) | 7 (6.4) | 64 (100) | < .001 |
| Mass spectrometry | 8 (4.7) | 6 (5.6) | 2 (3.2) | .712 |
| Scintigraphy | 165 (91.6) | 112 (96.6) | 53 (82.8) | < .001 |
| Perugini score of 2-3 | 125 (75.8) | 112 (100) | 13 (24.5) | < .001 |
| MGUS | 42 (41.2) | |||
| Hereditary ATTR | 18 (15.5) | |||
| Cardiac involvement | ||||
| NYHA class III-IV at diagnosis | 61 (33.9) | 28 (24.1) | 31 (51.5) | < .001 |
| History of HF admission | 106 (58.9) | 61 (52.6) | 45 (70.3) | .021 |
| History of atrial fibrillation | 69 (38.3) | 59 (50.9) | 10 (15.6) | < .001 |
| History of stroke | 18 (10.0) | 10 (8.6) | 8 (12.5) | .406 |
| Previous pacemaker implantation | 27 (15.0) | 24 (20.7) | 3 (4.7) | .004 |
| Previous cardioverter-defibrillator implantation | 3 (1.7) | 1 (0.9) | 2 (3.1) | .288 |
| Extracardiac involvement | ||||
| Carpal tunnel syndrome | 57 (31.7) | 48 (41.4) | 9 (14.1) | < .001 |
| Lumbar spinal stenosis | 11 (6.1) | 11 (9.5) | 0 | .008 |
| Macroglossia | 6 (3.3) | 2 (1.7) | 4 (6.3) | .188 |
| Peripheral neuropathy | 45 (25.0) | 25 (21.6) | 20 (31.3) | .150 |
| Dysautonomia | 95 (52.8) | 71 (61.2) | 24 (37.5) | .002 |
| Gastrointestinal involvement | 59 (32.8) | 34 (29.3) | 25 (39.1) | .182 |
| Renal involvement* | 116 (64.4) | 59 (50.9) | 57 (89.1) | < .001 |
AL, amyloid light-chain amyloidosis; ATTR, transthyretin amyloidosis; HF, heart failure; MGUS, monoclonal gammopathy of undetermined significance; NYHA, New York Heart Association functional class.
Values represent No. (%) or mean ± standard deviation.
Among patients with AL, 11 (17%) were in Mayo stage III at diagnosis and 48 (75%) in stage IV. Among patients with ATTR, 39 (34%) were in Gillmore stage II and 25 (22%) in stage III.15
In the electrocardiograms, one fourth of the series had atrial fibrillation at the first assessment, with a significantly higher rate for ATTR patients (36% vs 9%; P < .001). In the AL forms, the classic low-voltage pattern was found in 34 patients (54%) and the pseudoinfarct pattern in 32 (50%), which comprised the most frequent electrocardiographic pattern in the entire cohort (table 3).
Complementary baseline tests in patients with cardiac amyloidosis according to subtype
| Total (n = 180) | ATTR (n = 116) | AL (n = 64) | P | |
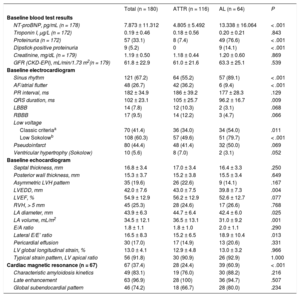
|---|---|---|---|---|
| Baseline blood test results | ||||
| NT-proBNP, pg/mL (n = 178) | 7.873 ± 11.312 | 4.805 ± 5.492 | 13.338 ± 16.064 | < .001 |
| Troponin I, μg/L (n = 172) | 0.19 ± 0.46 | 0.18 ± 0.56 | 0.20 ± 0.21 | .843 |
| Proteinuria (n = 172) | 57 (33.1) | 8 (7.4) | 49 (76.6) | < .001 |
| Dipstick-positive proteinuria | 9 (5.2) | 0 | 9 (14.1) | < .001 |
| Creatinine, mg/dL (n = 179) | 1.19 ± 0.50 | 1.18 ± 0.44 | 1.20 ± 0.60 | .869 |
| GFR (CKD-EPI), mL/min/1.73 m2(n = 179) | 61.8 ± 22.9 | 61.0 ± 21.6 | 63.3 ± 25.1 | .539 |
| Baseline electrocardiogram | ||||
| Sinus rhythm | 121 (67.2) | 64 (55.2) | 57 (89.1) | < .001 |
| AF/atrial flutter | 48 (26.7) | 42 (36.2) | 6 (9.4) | < .001 |
| PR interval, ms | 182 ± 34.9 | 186 ± 39.2 | 177 ± 28.3 | .129 |
| QRS duration, ms | 102 ± 23.1 | 105 ± 25.7 | 96.2 ± 16.7 | .009 |
| LBBB | 14 (7.8) | 12 (10.3) | 2 (3.1) | .068 |
| RBBB | 17 (9.5) | 14 (12.2) | 3 (4.7) | .066 |
| Low voltage | ||||
| Classic criteriaa | 70 (41.4) | 36 (34.0) | 34 (54.0) | .011 |
| Low Sokolowb | 108 (60.3) | 57 (49.6) | 51 (79.7) | < .001 |
| Pseudoinfarct | 80 (44.4) | 48 (41.4) | 32 (50.0) | .069 |
| Ventricular hypertrophy (Sokolow) | 10 (5.6) | 8 (7.0) | 2 (3.1) | .052 |
| Baseline echocardiogram | ||||
| Septal thickness, mm | 16.8 ± 3.4 | 17.0 ± 3.4 | 16.4 ± 3.3 | .250 |
| Posterior wall thickness, mm | 15.3 ± 3.7 | 15.2 ± 3.8 | 15.5 ± 3.4 | .649 |
| Asymmetric LVH pattern | 35 (19.6) | 26 (22.6) | 9 (14.1) | .167 |
| LVEDD, mm | 42.0 ± 7.6 | 43.0 ± 7.5 | 39.8 ± 7.3 | .004 |
| LVEF, % | 54.9 ± 12.9 | 56.2 ± 12.9 | 52.6 ± 12.7 | .077 |
| RVH, > 5 mm | 45 (25.3) | 28 (24.6) | 17 (26.6) | .768 |
| LA diameter, mm | 43.9 ± 6.3 | 44.7 ± 6.4 | 42.4 ± 6.0 | .025 |
| LA volume, mL/m2 | 34.5 ± 12.1 | 36.5 ± 13.1 | 31.0 ± 9.2 | .001 |
| E/A ratio | 1.8 ± 1.1 | 1.8 ± 1.0 | 2.0 ± 1.1 | .290 |
| Lateral E/E’ ratio | 16.5 ± 8.3 | 15.2 ± 6.5 | 18.9 ± 10.4 | .013 |
| Pericardial effusion | 30 (17.0) | 17 (14.9) | 13 (20.6) | .331 |
| LV global longitudinal strain, % | 13.0 ± 4.1 | 12.9 ± 4.8 | 13.0 ± 3.2 | .966 |
| Typical strain pattern, LV apical ratio | 56 (91.8) | 30 (90.9) | 26 (92.9) | 1.000 |
| Cardiac magnetic resonance (n = 67) | 67 (37.4) | 28 (24.4) | 39 (60.9) | < .001 |
| Characteristic amyloidosis kinetics | 49 (83.1) | 19 (76.0) | 30 (88.2) | .216 |
| Late enhancement | 63 (96.9) | 28 (100) | 36 (94.7) | .507 |
| Global subendocardial pattern | 46 (74.2) | 18 (66.7) | 28 (80.0) | .234 |
AF, atrial fibrillation; AL, amyloid light-chain amyloidosis; ATTR, transthyretin amyloidosis; CKD-EPI, Chronic Kidney Disease Epidemiology Collaboration; GFR, glomerular filtration rate; LA, left atrial; LBBB, left bundle branch block; LV, left ventricular; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; NT-proBNP, N-terminal pro-B-type natriuretic peptide; RBBB, right bundle branch block; RVH, right ventricular hypertrophy.
Values represent No. (%) or mean ± standard deviation.
Regarding the echocardiographic evaluation, the mean septal thickness of the cohort was 16.8 ± 3.4mm and 20% of the patients had eccentric LVH. No significant differences in LVEF were seen between ATTR and AL, although there was a tendency for worse LVEF in AL (56.2% ± 12.9% vs 52.6% ± 12.7%; P = .077). In contrast, the AL forms had lower left atrial dilatation (44.7 ± 6.4 vs 42.4 ± 6.0mm, P = .025; 36.5 ± 13.1 vs 31.0 ± 9.2mL/m2, P = .001) but a significantly higher lateral E/E’ ratio (15.2 ± 6.5 vs 18.9 ± 10.4cm/s; P = .013).
Of the 67 patients who underwent cardiac magnetic resonance (37%), more than 80% showed the characteristic gadolinium kinetics comprising faster myocardial nulling vs that of the blood pool. In addition, late gadolinium enhancement was seen in almost the entire series (63; 96.9%), with a global subendocardial pattern in more than half of the patients (18; 67%), particularly in the AL forms (28; 80%).
During follow-up, 9 patients (6%) required pacemaker implantation (6 ATTR patients and 3 AL); 2 patients underwent a defibrillator implantation in primary prevention (one to a patient with AL and restrictive phenotype who received a resynchronization device in another center after documentation of complete block and another to a patient with ATTRwt with severe ventricular dysfunction and syncope).
Extracardiac involvementThere was clinical evidence of amyloid deposition in the connective tissue in more than half of the patients with ATTR, mainly in the form of carpal tunnel syndrome (48 patients; 41%) or lumbar spinal stenosis (11; 10%). AL was also associated with carpal tunnel syndrome, although at a lower frequency (14%; P < .001). Neurological and gastrointestinal (vomiting and diarrhea) symptoms were relatively frequent (25% and 33%, respectively). Neurological involvement was evident in 13 of the 18 patients with hATTR (72%); peripheral neuropathy symptoms were also found in some of the ATTRwt group (12%).
Targeted therapy and transplantationDuring the study period, 18 patients (10%) participated in some type of clinical trial with specific antiamyloid molecules.
In this period, 15 patients with ATTR (13%) received some type of targeted therapy. Most (12 patients) received a stabilizing therapy (tafamidis in 10 patients and diflunisal in 2). Three patients with hATTR and neurological involvement were treated with gene silencing (patisiran) designed to suppress TTR production after disease progression during stabilizing therapy (2 patients) or from onset (1 patient). Finally, 2 patients underwent treatment with epigallocatechin-3-gallate, a polyphenol from green tea.
Most patients with AL (72%) received antineoplastic agents, 44 with bortezomib combinations and 2 with alkylating agents. Six patients underwent autologous hematopoietic cell transplantation after a previous treatment, and 18 were unable to receive treatment and received just a single cycle due to their death.
During the period analyzed, no patient with hATTR underwent liver transplantation. In contrast, 3 patients with AL underwent heart transplantation (table 4).
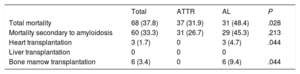
Mortality and transplantation in cardiac amyloidosis patients according to subtype
| Total | ATTR | AL | P | |
|---|---|---|---|---|
| Total mortality | 68 (37.8) | 37 (31.9) | 31 (48.4) | .028 |
| Mortality secondary to amyloidosis | 60 (33.3) | 31 (26.7) | 29 (45.3) | .213 |
| Heart transplantation | 3 (1.7) | 0 | 3 (4.7) | .044 |
| Liver transplantation | 0 | 0 | 0 | |
| Bone marrow transplantation | 6 (3.4) | 0 | 6 (9.4) | .044 |
AL, amyloid light-chain amyloidosis; ATTR, transthyretin amyloidosis.
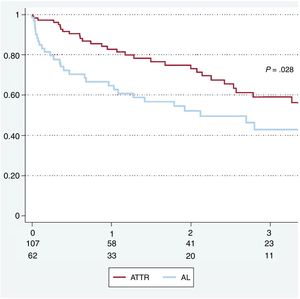
During a mean follow-up of 1.6 ± 1.7 years, the total mortality of the cohort was 38% (68 patients); it was significantly higher in AL patients (48% vs 32% in ATTR; P = .028) (table 4). In 60 of those who died (88%), the cause of death was attributable to the amyloidosis.
The overall 1-year survival was 76%. The 12-, 24-, and 36-month survival rates were 65%, 52%, and 43% in AL patients and 83%, 73%, and 59% in ATTR patients, respectively (figure 3).

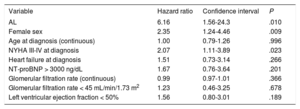
In the multivariable analysis, the only factors significantly associated with death or heart transplantation were AL subtype (hazard ratio [HR] = 6.16; 95% confidence interval [95%CI], 1.56-24.3; P = .010), female sex (HR = 2.35; 95%CI, 1.24-4.46; P = .009), and NYHA class III-IV at diagnosis (HR = 2.07; 95%CI, 1.11-3.89; P = .023) (table 5).
Multivariable analysis of factors predicting mortality in 180 patients with cardiac amyloidosis
| Variable | Hazard ratio | Confidence interval | P |
|---|---|---|---|
| AL | 6.16 | 1.56-24.3 | .010 |
| Female sex | 2.35 | 1.24-4.46 | .009 |
| Age at diagnosis (continuous) | 1.00 | 0.79-1.26 | .996 |
| NYHA III-IV at diagnosis | 2.07 | 1.11-3.89 | .023 |
| Heart failure at diagnosis | 1.51 | 0.73-3.14 | .266 |
| NT-proBNP > 3000 ng/dL | 1.67 | 0.76-3.64 | .201 |
| Glomerular filtration rate (continuous) | 0.99 | 0.97-1.01 | .366 |
| Glomerular filtration rate < 45 mL/min/1.73 m2 | 1.23 | 0.46-3.25 | .678 |
| Left ventricular ejection fraction < 50% | 1.56 | 0.80-3.01 | .189 |
AL, amyloid light-chain amyloidosis; NT-proBNP, N-terminal pro-B-type natriuretic peptide; NYHA, New York Heart Association functional class.
This study describes the characteristics of a large cohort of patients with CA. Our analysis of the clinical practice of a center experienced in the treatment and follow-up of this condition demonstrate that ATTR is the most frequent form of CA in Spain. The data reveal the various presentations of CA, as well as the heterogeneous electrocardiographic and echocardiographic manifestations. Our work also shows the high yield of EMB in the AL forms and the recent increase in the number of patients noninvasively diagnosed with ATTR. Finally, AL subtype, advanced functional class, and female sex were identified to be predictors of mortality or transplantation.
Few published series have described the characteristics of patients with CA.16,17 Interest in CA has grown in recent years with the publication of a large number of studies in this field. Our data are the fruit of the experience of a center with a heart transplantation program and a congenital heart disease unit that is a pioneer in the management of these patients, as well as the result of continuous improvements in the understanding of the disease.1,2,18–25
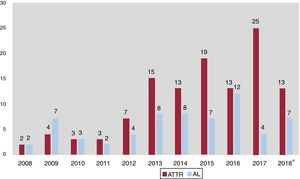
AL was previously considered the most common form of CA in developed countries, with ATTR a subgroup poorly represented in previous publications. However, current data indicate that ATTR is actually the most frequent CA5 and has thus far been underdiagnosed, with an unknown true prevalence. ATTR patients comprise more than half of CA patients treated in our center and there has been a change in trends in recent years, with a clear increase in these patients (figure 4). The possibility of specific therapies for the treatment of ATTR2 makes it even more important to identify these patients and will probably help to increase the number of patients diagnosed with CA, both ATTR and AL.

The diagnosis of CA continues to be a key issue. Our data show that the classic clinical profile—restrictive cardiomyopathy with concentric hypertrophy and low ECG voltage—is found in only a minority of patients. Although HF is the most frequent manifestation, the clinical profile of ATTR is much more varied than that of the AL forms. This finding requires a change in mentality when this condition is suspected. The mean time from symptom onset to diagnosis in patients treated in our center was 0.6 ± 0.7 years for AL and 2.8 ± 4.3 years for ATTR. The mean diagnostic delay in both groups was less than that reported in the literature, a delay of about 3 years for ATTR forms with less than half of patients with AL diagnosed in the first 6 months.6 We believe that the reduced delay in our patients is a consequence of the high clinical suspicion, the early performance of EMB, and the elevated number of patients noninvasively diagnosed with ATTR.
In our opinion, the experience accumulated from more than 20 000 bone 99mTc-DPD scintigraphies in the last 10 years supports the systematic incorporation of this test into the diagnostic process for ATTR-CA. Indeed, 72% of our patients with ATTR were noninvasively diagnosed in our series. In addition, magnetic resonance, with its strong capacity for morphological, functional, and tissue characterization, is fundamental for the noninvasive diagnosis of CA.19 Its ability to evaluate parameters such as extracellular volume and T1 mapping will probably increase its diagnostic yield in the coming years.26
However, histological evidence continues to be fundamental in a considerable number of patients, particularly those suspected to have AL. We have already reported the importance of targeting the affected organ without delay8 and the current data demonstrate the high yield of EMB.
Once a patient is diagnosed with CA, it is critical to determine the subtype. This differentiation is not always easy: not all uptake on scintigraphy reflects ATTR and not all monoclonal component in CA indicates AL. We found a significant prevalence of monoclonal gammopathy of undetermined significance in the ATTR population (41%), higher than in previous cohorts and vs the same-age general population. In this context, the use of immunohistochemical techniques and proteomic analysis is fundamental. In our case, mass spectrometry was necessary to determine the amyloidosis subtype in 5% of biopsied patients.
Finally, our data show a 1-year survival of 76%, comparable to that of other referral centers,16 and worse prognosis for AL vs ATTR (1-year survival of 65% vs 83%). These is a general consensus on the usefulness of the new antimyeloma drugs (proteasome inhibitors, immunomodulators, and monoclonal antibodies) for the treatment of patients with AL and that high chemotherapy doses and autologous transplantation can achieve long-lasting remission in selected patients.10 The poor prognosis of patients with AL and the complexity of the required treatments support the need for these patients to be treated by specialized teams that can benefit from close collaboration with advanced HF and hematology units.
In light of our results, we must stress that CA continues to be a condition with poor prognosis, even in referral centers. We believe that the complexity of the diagnosis and management of patients with ATTR and AL, as well as the possibility of diverse specific treatments, support the creation of referral centers that can apply their resources and experience to best manage the needs of these patients, similar to other countries in Europe.27
LimitationsThe present data were derived from a descriptive and retrospective study performed in a single center and subject to selection and survival biases. Notably, about 80% of the patients were diagnosed in the last 5 years, which could also have influenced the results.
CONCLUSIONSATTR is the most frequent form of CA. The clinical presentation and characteristics of CA patients are heterogeneous and differ between ATTR and AL. Although EMB is associated with high diagnostic yield and few complications, ATTR can now be noninvasively diagnosed in a considerable number of patients. AL subtype, female sex, and NYHA class III-IV at diagnosis are independently associated with worse prognosis. Given the development of new treatments for CA, cardiologists need to familiarize themselves with the characteristics and diagnostic process for these patients.
CONFLICTS OF INTERESTE. Gonzalez-Lopez has received honoraria for presentations and advisory activity from Pfizer and Proclara. I. Krsnik has received honoraria for presentations from Janssen and for advisory activity from Celgene and Prothena, as well as research funding from Prothena. P. Garcia-Pavia has received honoraria for presentations or advisory activity from Akcea, Alnylam, Eidos, Neuroinmmune, Pfizer, and Prothena and research funding from Alnylam, Pfizer, and Prothena.
- –
CA is an underdiagnosed condition with a considerable diagnostic delay.
- –
In recent years, numerous advances have been made in the possible noninvasive diagnosis of the condition and in the development of new therapies.
- –
ATTR is the most frequent form of CA in Spain.
- –
The clinical presentation and characteristics of CA patients are heterogeneous and differ substantially between ATTR and AL.
- –
In Spain, the diagnosis of ATTR-CA is increasingly noninvasive.
- –
AL subtype, female sex, and NYHA class III-IV at diagnosis are independently associated with worse prognosis.
To Ariadna González and Almudena Ariza, for their work in patient coordination and management of the Amiloidosis.es portal of Hospital Puerta de Hierro Majadahonda.
To Drs Lucía Galán and Antonio Guerrero of the Neurology Service of Hospital Clínico San Carlos, Madrid, for their efforts to care for patients with hATTR.
Supplementary data associated with this article can be found in the online version available at https://doi.org/10.1016/j.rec.2019.12.020