Common arterial trunk is a truncoconal congenital heart disease that represents up to 4% of all congenital heart disease and is characterized by a single arterial trunk from which the coronary, pulmonary, and systemic arteries originate. The most widely-used classification is that of Collett and Edwards, based on the anatomic origin of the pulmonary arteries. Without surgical treatment, 80% of patients die prematurely, and survival to adolescence or adulthood is rare; almost all survivors develop severe vascular obstructive disease or Eisenmenger syndrome.1,2
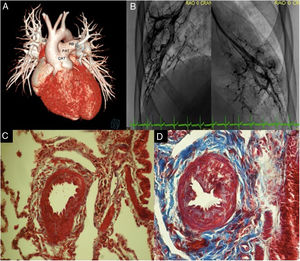
With prior ethics committee authorization and consent from the patient's parents, we present the case of a 15-year-old male patient—diagnosed at 2 months of age with common arterial trunk type 1 that was not surgically treated based on the parents’ decision—who was reassessed due to onset of dyspnea. On admission, pulse oximetry was 85% and there was a left parasternal systolic murmur. Transthoracic echocardiography confirmed the prior diagnosis, showing a tri-leaflet truncal valve with moderate regurgitation and a ventricular septal defect measuring 20×26mm. This was followed by computed tomography which showed abnormal origin of the left coronary artery (figure 1A). Catheterization showed systemic pulmonary pressure, with end-diastolic pressure of the left and right ventricles of 15mmHg. On calculation using the Fick method with an assumed oxygen consumption of 132mL/min/m2, Qp/Qs was 4.7, with a pulmonary vascular resistance index of 5.45 UW/m2 and PVR/SVR ratio=0.19; when oxygen was given, the Qp/Qs ratio increased to 5.08 and the pulmonary vascular resistance index decreased to 4.49 UW/m2 (table 1); a homogenous right capillary blush was observed (figure 1B). Karyotype study and fluorescent in situ hybridization (FISH) were reported as normal.
; Heath and Edwards grade I. D: left pulmonary artery biopsy, with vascular wall thickening due to smooth muscle (red) and intimal proliferation, with fibrous tissue (blue); Heath and Edwards grade II. CAT, common arterial trunk; LB, left pulmonary branch; RB, right pulmonary branch; PAT, pulmonary arterial trunk.")
A: 3D reconstruction. Common arterial trunk type I. B: magnification pulmonary wedge angiography with right and left capillary blush. C: Masson stain. Right pulmonary artery biopsy, with proliferation of the wall due to smooth muscle hyperplasia (red); Heath and Edwards grade I. D: left pulmonary artery biopsy, with vascular wall thickening due to smooth muscle (red) and intimal proliferation, with fibrous tissue (blue); Heath and Edwards grade II. CAT, common arterial trunk; LB, left pulmonary branch; RB, right pulmonary branch; PAT, pulmonary arterial trunk.
Cardiac catheterization with 21% FiO2 before surgery and 23 months after surgery
| SaO2, preoperative and postoperative, % | Systolic pressure, preoperative and postoperative, mmHg | Diastolic pressure, preoperative and postoperative, mmHg | End-diastolic pressure, preoperative and postoperative, mmHg | Mean pressure, preoperative and postoperative, mmHg | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| SVC | 54 | 70 | ||||||||
| IVC | 67 | 81 | ||||||||
| RV | 85 | 61 | 15 | 8 | ||||||
| PAT | 90 | 78 | 85 | 52 | 35 | 9 | 60 | 28 | ||
| LV | 95 | 95 | 99 | 100 | 15 | 11 | ||||
| DA | 95 | 95 | 99 | 100 | 44 | 53 | 63 | 85 | ||
| MVS | 57,25 | 72,75 | ||||||||
DA, descending aorta; IVC, inferior vena cava; LF, left ventricle; MVS, mixed venous saturation; PAT, pulmonary arterial trunk; RV, right ventricle; SaO2, oxygen saturation; SVC, superior vena cava.
It was decided to perform surgical correction, with connection of the pulmonary ventricle using a 22-mm woven Dacron tube, implantation of a 21-mm bovine aortic valve prosthesis in the pulmonary position, closure of the ventricular septal defect with a bovine pericardial patch, and aortic valvuloplasty. Right and left pulmonary biopsy reported muscularization of the tunica media of the intralobular and centrilobular arteries, with no intimal reaction, small vessels with fibrin clots, and hemosiderin-containing alveolar macrophages (figure 1C,D).
The patient progressed well, in NYHA functional class I. Follow-up catheterization at 23 months found a mean pulmonary pressure of 28mmHg, pulmonary vascular resistance index of 4.92 UW/m2, and PVR/SVR ratio=0.19.
Left to the natural history of the disease, it is rare for patients to survive to adolescence and adulthood; almost all develop severe pulmonary vascular obstructive disease or Eisenmenger syndrome.2 The pathophysiological mechanisms that determine the reversible or irreversible nature of pulmonary hypertension in congenital heart disease remain unclear. Blood flow and pressure are key triggers for pulmonary vascular remodeling in congenital heart disease: with greater flow and pressure, the blood flow in the whole pulmonary arterial bed is disrupted, causing inflammation and proliferation.3
Genetic susceptibility factors have been identified that may predispose to or accelerate pulmonary vascular remodeling: the most noteworthy are bone morphogenetic protein receptor type 2 (BMPR2) and transcription factor Sox17.4 Roberts et al.5 identified mutations in 6% of patients with congenital heart disease-associated pulmonary hypertension. In patients with congenital heart disease, Liu et al.6 found a significant difference in BMPR2 mutation between those with and those without pulmonary vascular disease.
The case presented here had an unusual natural history. The patient did not have protective anatomical abnormalities (pulmonary branch hypoplasia/stenosis) or pulmonary banding; furthermore, he lived in a city that has an altitude of 2240 m, and did not develop early irreversible pulmonary hypertension despite a large shunt—this could be related to physiological and genetic adaptation processes that have been described in animals living at high altitude. It is unknown if protective genetic factors exist that would explain why some patients, despite having congenital heart disease with a large shunt, develop pulmonary hypertension later and to a lesser severity.
In conclusion, patients should be assessed thoroughly as candidates for surgical correction without using age as an exclusion criterion for corrective treatment; it is important to understand the mechanisms that allow some patients to be protected and not develop pulmonary vascular disease.
FUNDINGNo funding was received for this article.
AUTHORS’ CONTRIBUTIONSConcept/design: J. Calderón-Colmenero, J. A. García-Montes, J. L. Cervantes-Salazar. Writing: A. Aranda-Frausto, F. Castillo-Castellón, E. Lupinta-Paredes. Critical review/approval: J. Calderón-Colmenero, J. A. García-Montes, A. Aranda-Frausto, F. Castillo-Castellón, E. Lupinta-Paredes, J. L. Cervantes-Salazar.
CONFLICTS OF INTERESTThe authors declare no conflicts of interest.