El tratamiento convencional con ácido acetilsalicílico y clopidogrel (una tienopiridina) ha quedado superado por el uso de nuevos inhibidores del receptor P2Y12 como prasugrel y ticagrelor. El prasugrel, al igual que toda tienopiridina, se administra oralmente e inhibe de manera irreversible el receptor, pero, pese a ser un profármaco, tiene un metabolismo más rápido y eficaz que el clopidogrel, lo que deriva en un mayor beneficio clínico. El ticagrelor (derivado nucleósido) proporciona un bloqueo reversible del receptor y no requiere bioactivación hepática, con lo que ofrece una rápida y potente inhibición plaquetaria. Además, el cangrelor y el elinogrel son dos compuestos en fase de desarrollo disponibles como formulaciones intravenosas que presentan un efecto directo, reversible y de mayor seguridad para los pacientes sometidos a cirugía coronaria. En los últimos años, de estos compuestos, y muy especialmente del ticagrelor, se han demostrado efectos beneficiosos más allá de la inhibición plaquetaria, los llamados efectos pleiotrópicos.
Palabras clave
La aterosclerosis y sus complicaciones isquémicas cardiovasculares son las causas más comunes de muerte y discapacidad en todo el mundo1. De hecho, la Organización Mundial de la Salud informó en 2010 que la enfermedad cardiovascular representa alrededor del 30,5% de todas las muertes en el mundo y estima que en 2030 más de 23,3 millones de personas morirán cada año por enfermedades cardiovasculares2, 3, 4. La trombosis desempeña un papel primordial en la ocurrencia de eventos isquémicos coronarios (p. ej., síndrome coronario agudo [SCA]) y se desencadena tras la erosión o rotura de las placas ateroscleróticas de alto riesgo ricas en lípidos5,6. La exposición de estructuras vasculares o componentes de núcleo necrótico de la placa aterosclerótica a la circulación sanguínea desencadena la activación del factor tisular y la posterior formación de una monocapa de fibrina (activación de la cascada de la coagulación), y simultáneamente induce el reclutamiento de las plaquetas circulantes y las células inflamatorias al lugar de lesión7,8. En la hemostasia, la interacción del factor de von Willebrand (FVW) circulante a través de su dominio A3, con el colágeno expuesto, constituye el paso inicial en el proceso de adhesión plaquetaria, al proporcionar un punto de anclaje para las plaquetas a través de la interacción del receptor plaquetario glucoproteína (GP) Ib-IX-V con el dominio A1 del FVW9. Este primer contacto plaqueta-vaso se ve reforzado por la interacción directa del colágeno con los receptores plaquetarios GPIa y la GPVI, lo que deriva en la consiguiente activación de la plaqueta.
Una vez activadas, las plaquetas liberan difosfato de adenosina (ADP) y tromboxano A2 (TXA2), entre otros agonistas, lo que perpetúa el proceso7,8,10. El ADP puede unirse a dos receptores purinérgicos acoplados a proteínas G presentes en la superficie de la plaqueta, el receptor P2Y1 y el receptor P2Y1211,12. Por un lado, la unión del ADP con el receptor P2Y1 deriva en un incremento en la concentración intracelular de calcio, así como en un cambio conformacional de las plaquetas. Esto conlleva una respuesta débil y transitoria de agregación plaquetaria. Por el contrario, la interacción del ADP con el receptor plaquetario P2Y12, además de perpetuar el incremento de calcio intracelular, disminuye la cantidad de monofosfato cíclico de adenosina (AMPc), con la consiguiente agregación plaquetaria irreversible (Figura 1)11. El TXA2 se produce por la activación de la enzima fosfolipasa A2 y la resultante liberación de ácido araquidónico de la membrana de la plaqueta (Figura 1). El ácido araquidónico liberado interacciona con la enzima ciclooxigenasa 1, lo que da lugar a la formación TXA2, que a su vez perpetúa el incremento del calcio intracelular al interaccionar con el receptor del TXA2 (Figura 1). Finalmente, la trombina, sintetizada en el plasma tras la activación de la cascada de coagulación, interacciona con el receptor de la trombina activado por proteasa tipo 1 (PAR-1) en la superficie de la plaqueta, lo que se traduce en un aumento de calcio y la activación de la fosfolipasa C (Figura 1)13. Todo ello deriva en el cambio conformacional y la consiguiente activación del receptor plaquetario GPIIb/IIIa, el receptor plaquetario más abundante (Figura 1). Una vez activado, este receptor reconoce y se une a unas secuencias determinadas de aminoácidos (Arg-Gly-Asp, también llamada RGD, o Lys-Gln-Ala-Gly-Asp-Val, también llamada KQAGDV) presentes en grandes cantidades en el fibrinógeno circulante. Cabe destacar que, aunque en menor medida, la secuencia Arg-Gly-Asp también está contenida en el FVW, la fibronectina y la vitronectina, sustancias que pueden actuar también como ligandos del receptor GPIIb/IIIa (Figura 1). La unión del fibrinógeno circulante con el receptor GPIIb/IIIa da lugar a la formación de puentes entre las plaquetas activadas que inducen la formación de agregados plaquetarios7.

Figura 1. Mecanismos de activación y agregación plaquetarias y posibles dianas terapéuticas. La interacción del difosfato de adenosina (ADP) o trombina con sus receptores plaquetarios y la activación de la fosfolipasa A2 (PLA2), con la consiguiente formación de tromboxano A2 (TXA2), derivan en la activación de señalización intraplaquetaria y en incremento del calcio intracelular, que desencadenan la activación de la glucoproteína IIb/IIIa, receptor plaquetario clave en la agregación plaquetaria. Se han desarrollado diversos fármacos capaces de bloquear los receptores diana y la síntesis de TXA2. AAS: ácido acetilsalicílico.
Teniendo presente el papel clave en la formación del trombo del ADP, la síntesis de TXA2 y la trombina, así como la activación de la GPIIb/IIIa, los esfuerzos se han centrado en la búsqueda de fármacos capaces de bloquear o inhibir sus vías de señalización de manera eficaz intentando evitar las hemorragias (Figura 1)14,15. Concretamente, en los últimos años ha habido un auge en el desarrollo de inhibidores del receptor P2Y12.
Esta revisión se centra en la vía de actuación del ácido acetilsalicílico (AAS), los inhibidores de la GPIIb/IIIa y la trombina y los mecanismos de acción y las propiedades de los distintos inhibidores del receptor P2Y12.
Ácido acetilsalicílicoEl AAS fue el primer fármaco antiplaquetario empleado en clínica16. A través de la acetilación de la serina-529, inhibe de manera irreversible la ciclooxigenasa 1 plaquetaria y previene la conversión del ácido araquidónico en prostaglandina G2/H2 y su consiguiente metabolismo por la enzima tromboxano sintetasa en TXA2 (Figura 1)17. Además de inhibir la síntesis de TXA2, diversos estudios indican que el tratamiento con AAS conlleva una reducción de los factores plaquetarios 3 y 4, disminuye los factores de coagulación II, VII, IX y X (en dosis ≥ 200 mg) y presenta cierta actividad fibrinolítica (en dosis de 1.800 mg)18. El AAS es el fármaco más ampliamente utilizado en prevención secundaria, pero solo produce una reducción del 22% del riesgo relativo de sufrir nuevos accidentes isquémicos y deja una elevada proporción de pacientes en riesgo19. De ahí el interés del tratamiento combinado con otros antiplaquetarios a fin de bloquear más eficazmente las plaquetas14.
Inhibidores del receptor de la glucoproteína IIIb/IIIaLos anti-GPIIb/IIa representan una diana ideal para la prevención del SCA, ya que bloquean la parte final de la formación del trombo sea cual fuere el estímulo que desencadenara su activación inicial. El interés que inicialmente despertó el posible uso oral de los inhibidores de la GPIIb/IIIa se vio afectado por los resultados negativos obtenidos en diversos estudios en pacientes con SCA o sometidos a intervención percutánea en los que se evidenció un aumento en la mortalidad20. A pesar de que se desconoce las causas, se propusieron posibles explicaciones que incluían la ausencia de tratamiento combinado con AAS, un incremento en la expresión de moléculas de adhesión plaqueta/leucocito, un grado insuficiente de inhibición plaquetaria, la presencia de polimorfismos genéticos y la inducción de apoptosis miocárdica vía estimulación de caspasa-321. Por el contrario, se ha demostrado la utilidad clínica de los compuestos formulados para administrar por vía intravenosa asociada al intervencionismo coronario, especialmente en pacientes de alto riesgo22. Estos fármacos pueden clasificarse según su mecanismo de acción en inhibidores irreversibles de la GPIIb/IIIa (abciximab) y los que los inhiben de manera competitiva y reversible en el lugar de unión para la secuencia RGD (eptifibatida, tirofibán y lamifibán) (Figura 1).
Inhibidor irreversibleEl abciximab proviene del fragmento Fab del anticuerpo monoclonal de origen murino (7E3), posteriormente quimerizado para reducir su inmunogenicidad24. El abciximab se une de modo inespecífico e irreversible al receptor plaquetario GPIIb/IIIa e impide la unión del fibrinógeno y otras moléculas de adhesión que derivarían en la formación de la agregación plaquetaria (Figura 1). A diferencia de los otros anti-GPIIb/IIIa, el abciximab no es específico para este receptor y puede unirse también al receptor la vitronectina (αvβ3) y al receptor leucocitario Mac-1.
El abciximab produce una inhibición plaquetaria dependiente de la dosis25. Tras un bolo endovenoso de 0,25 mg/kg, se observa una reducción de la función plaquetaria prácticamente completa a los 10 min, que corresponde a una ocupación de más del 80% de los receptores. Para mantener este grado de inhibición, se requiere una infusión continua ajustada al peso del paciente. La vida media de abciximab es larga, entre 6 y 12 h. Sin embargo, debido a su unión irreversible con el receptor, la recuperación de la función plaquetaria después de interrumpir la infusión es gradual, y ocurre durante los 4–6 días posteriores al tratamiento. Por ello, para revertir sus efectos antiplaquetarios, es necesaria la transfusión de plaquetas. Entre los efectos secundarios de este fármaco, además del riesgo hemorrágico asociado al gran potencial inhibidor, destacan su capacidad inmunogénica (a pesar de ser inferior a la descrita de los anticuerpos murinos) y la trombocitopenia.
Inhibidores reversiblesEstos inhibidores se han desarrollado sintéticamente con base en su similitud con la secuencia de reconocimiento RGD que existe en el fibrinógeno. En este contexto, la estructura de eptifibatida se basa en la secuencia KGD, en vez de la RGD, en la que se sustituye una arginina por una lisina. Otro enfoque también de tipo competitivo adoptado en el diseño de estos fármacos es imitar la secuencia RGD sintetizando pequeñas moléculas de derivados peptídicos (con enlaces peptídicos) y no peptídicos (sin dichos enlaces), con el fin de superar los problemas de inestabilidad y corta vida de los péptidos sintéticos. Ejemplos de derivados del péptido RGD son el lamifibán, que es un peptidomimético, y el tirofibán, derivado no peptídico. Todos poseen una rápida acción antiplaquetaria (pocos minutos tras su infusión) y una vida media corta (2–3 h) y, debido a la reversibilidad de su efecto, la agregación plaquetaria vuelve a > 50% del valor basal tras 4 h de suspensión del tratamiento25.
Bloqueadores del receptor de la trombinaActualmente existen dos inhibidores del receptor de PAR-1, vorapaxar (SCH 530348) y atopaxar (E5555), ambos aún en fase de desarrollo clínico (fase III)26. Los dos se administran por vía oral y son moléculas no proteicas que actúan como potentes antagonistas competitivos del receptor PAR-1 de la trombina. El vorapaxar se absorbe rápidamente y muestra mayor afinidad para PAR-1 que el atopaxar. De hecho, alcanza la máxima acción antiplaquetaria a las 2 h, mientras que el atopaxar es algo más tardío (3,5 h)27. El varopaxar tiene una vida media larga, estimada en hasta 311 h, mientras que la del atopaxar es de solo 23 h. El vorapaxar y el atopaxar presentan una metabolización hepática lenta por el citocromo CYP3A4. Por lo tanto, la coadministración de fármacos que modifican la actividad metabólica de la enzima (p. ej., ketoconazol, rifampicina) puede modular la concentración final del vorapaxar. La eliminación de ambos fármacos es principalmente por vía fecal (95%), aunque un 5% se excreta por vía renal.
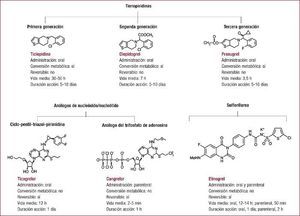
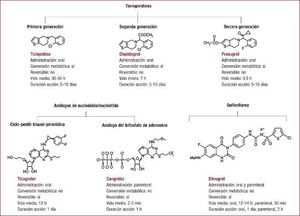
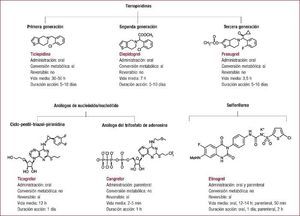
Inhibidores del receptor P2Y12Propiedades antiplaquetariasTienopiridinasEn 1991 se aprobó el uso del primer antagonista del receptor P2Y12, la ticlopidina, una tienopiridina. Desde entonces se han desarrollado e introducido en la práctica clínica dos tienopiridinas más, el clopidogrel, tienopiridina de segunda generación, y el prasugrel, de tercera generación15,28,29. Las tienopiridinas se administran oralmente, son profármacos y derivan en la inhibición irreversible del receptor P2Y12 (Figura 2).

Figura 2. Clasificación y propiedades de los distintos inhibidores del receptor del difosfato de adenosina P2Y12.
Ticlopidina y clopidogrelLa ticlopidina bloquea de manera irreversible el receptor P2Y12 una vez biotransformada en el hígado, mediante la actuación de CYP2C19 y CYP2B6, en oxoticlopidina y finalmente en su forma tiolactona (activa)30. Su vida media es de 30–50 h, aunque su máxima actividad antiplaquetaria se detecta entre 3 y 5 días tras su administración31 (Figura 2). A pesar de su eficacia como un antagonista del receptor de ADP, el amplio uso de la ticlopidina se vio significativamente afectado por la incidencia, grave aunque poco frecuente, de trombocitopenia y neutropenia32,33. El clopidogrel reemplazó gradualmente a la ticlopidina y se introdujo como una tienopiridina de segunda generación sin toxicidad. Al igual que la ticlopidina, el clopidogrel inhibe de manera irreversible (unión covalente) el receptor P2Y12 y es un profármaco. El metabolismo del clopidogrel implica dos vías hepáticas: una vía convierte la mayor parte del clopidogrel (hasta un 75%) en metabolitos ácidos inactivos mediante su desesterificación, mediada por una carboxilesterasa; la segunda vía consta de dos pasos hepáticos: el clopidogrel primero se convierte en 2-oxo-clopidogrel mediante la acción de las enzimas CYP2C19, CYP1A2 y CYP2B6 y, a continuación, en metabolito activo mediante CYP2B6, CYP3A, CYP2C9 y CYP2C19. La forma 2-oxo-clopidogrel también puede metabolizarse a su forma inactiva mediante la actuación de las esterasas. La excreción del clopidogrel se produce por vía renal (50%) y biliar (46%). Que el clopidogrel sea sustrato de las enzimas CYP2C y CYP2C19, respectivamente, conlleva un riesgo de interacciones farmacológicas34 y variaciones genéticas35, lo que resulta en una inhibición plaquetaria impredecible e insuficiente en ciertos pacientes35. La vida media del clopidogrel es de aproximadamente 7 h y su efecto antiagregante es máximo a las 2–3 h de administrarse una dosis de carga de 600 mg. Al igual que la ticlopidina, su irreversibilidad conlleva que su efecto antiplaquetario dure toda la vida de la plaqueta (unos 5–10 días) (Figura 2), lo que incrementa el riesgo de hemorragia y transfusión en caso de precisarse cirugía cardiotorácica36. Estas limitaciones han incentivado la búsqueda de nuevos agentes antiplaquetarios, a pesar de que múltiples estudios en diferentes entornos clínicos han demostrado que la administración de clopidogrel, solo o añadido a AAS, es superior a la monoterapia con AAS37, 38, 39.
PrasugrelEl prasugrel es una tienopiridina de tercera generación que, como tal, inhibe de manera irreversible el receptor P2Y12 (Figura 2)40. Por ello, su actividad antiplaquetaria también dura 5–10 días y se necesita interrumpir su administración 1 semana antes de la cirugía. Tras su absorción, el prasugrel se hidroliza por la acción de la carboxilesterasa a la tiolactona R-95913, un compuesto intermedio que después se metaboliza al metabolito activo R-138727 mediante CYP3A, CYP2B6, CYP2C9 y CYP2C19. En contraste con el clopidogrel, una esterasa, es esencial en la activación del profármaco más que en su inhibición, solo sufre un paso de metabolismo hepático y la conversión al metabolito activo se hace predominantemente a través de CPY3A4 y CYP2B6, por lo que no afecta a los pacientes con polimorfismos del CYP2C19. Que el prasugrel solo requiera un paso de metabolización hepática conlleva que tenga una respuesta de inhibición plaquetaria más rápida, constante y potente que el clopidogrel. De hecho, la vida media del prasugrel es de aproximadamente 3,5 h, pero su concentración máxima se alcanza a los 30 min de la administración. Además, el prasugrel ha mostrado más eficacia que el clopidogrel en pacientes con SCA41,42. Sin embargo, estos beneficios son a expensas de un aumento del riesgo de sangrado, lo que limita su uso en pacientes de 75 años o más, con un peso < 60 kg, con historia de accidente cerebrovascular o ataque isquémico transitorio o con posibilidad de someterse a cirugía cardiotorácica42,43. El prasugrel se excreta vía renal (68%) y fecal (27%).
Análogos de los nucleósidos/nucleótidosTeniendo en cuenta las limitaciones de las tienopiridinas, durante los últimos años se han desarrollado e investigado nuevos bloqueadores del receptor P2Y12: el ticagrelor, el cangrelor y el elinogrel15,29,44,45. El ticagrelor está aceptado para uso clínico, mientras que el cangrelor y el elinogrel están aún en fase de desarrollo clínico.
TicagrelorEl ticagrelor deriva de una nueva clase de agentes químicos, las ciclopentil-triazolopirimidinas (Figura 2)46. El ticagrelor, a diferencia de las tienopiridinas, se une reversiblemente y de manera no competitiva al receptor P2Y12; su vida media es de aproximadamente 12 h y su efecto antiagregante disminuye a las 48 h de la última dosis47. El ticagrelor no es un profármaco y por ello no requiere metabolismo hepático para su actividad, hecho que ofrece una ventaja farmacocinética importante sobre el clopidogrel y, en menor medida, el prasugrel. De hecho, el ticagrelor se absorbe rápidamente tras su administración oral y es biológicamente activo. Además, se metaboliza mediante la enzima CYP3A, genera un metabolito activo, el AR-C124910XX, presente en sangre a concentraciones de alrededor del 40% de la de ticagrelor, y presenta una potencia antiplaquetaria similar a la de su compuesto original48. Por ello, el ticagrelor resulta en una rápida acción antiplaquetaria (1,5–2 h tras su ingestión) más potente que la derivada de tomar clopidogrel48, 49, 50, 51. Su excreción es mayoritariamente biliar. Cabe destacar que, puesto que el ticagrelor se metaboliza vía CYP3A, se limita su administración a pacientes que reciben simvastatina y lovastatina en dosis > 40 mg. Es más, un estudio reciente ha señalado que se reduce la eficacia del ticagrelor si se administra con una dosis de AAS > 100 mg/día. Sin embargo, esta contraindicación está aún por contrastar.
CangrelorEl cangrelor es un análogo del ATP, se administra por vía endovenosa y es un inhibidor directo, reversible y competitivo del receptor P2Y12 (Figura 2). El cangrelor tiene un inicio rápido y una vida media corta, de aproximadamente 2–5 min. No requiere conversión hepática (se metaboliza en el plasma) y tiene un perfil farmacocinético dependiente de la dosis. La respuesta plaquetaria se recupera transcurridas 1–2 h tras su infusión. Desafortunadamente, dos grandes ensayos clínicos en fase III en pacientes sometidos a intervención coronaria percutánea no han demostrado su superioridad en comparación con el clopidogrel52,53. Sin embargo, a pesar de la falta de impacto clínico, la posibilidad de administrarlo vía parenteral y su reversibilidad lo hacen un candidato atractivo y fiable para pacientes de riesgo.
Derivado de la sulfonilureaElinogrelEl elinogrel pertenece a una clase única de antiagregantes plaquetarios conocidos como sulfonilureas54 (Figura 2). El elinogrel es una molécula pequeña que inhibe de manera directa y reversible el receptor P2Y12. Este antiplaquetario, a diferencia de todos los anteriores, puede administrarse por vía oral o intravenosa. La formulación oral del elinogrel tiene una vida media de 11–12 h y se excreta sin cambios, principalmente en orina (56%) y heces (48%)54. La formulación endovenosa tiene su inhibición plaquetaria máxima a los 20 min y las plaquetas recuperan la totalidad de su función transcurridas 24 h.
Propiedades extraplaquetarias de los inhibidores de los receptores P2Y12Las plaquetas no solo tienen un papel crucial en la formación del trombo, sino que además son una importante reserva de mediadores de inflamación. De ahí que inhibir la función plaquetaria pueda derivar en una disminución de ciertos marcadores inflamatorios, especialmente los asociados con plaquetas activadas, tales como el ligando de CD40 (CD40L) y la P-selectina6,8. Sin embargo, los antagonistas del receptor P2Y12 también han mostrado su capacidad de modular la respuesta inflamatoria inhibiendo las interacciones de las plaquetas con los leucocitos y de interaccionar directamente con las células blancas circulantes, con lo que se atenúa su activación. De hecho, se ha descrito la capacidad del clopidogrel de reducir las concentraciones séricas de CD40L, CRP y P-selectina y la formación de agregados de plaquetas-leucocitos55. En este contexto, durante los últimos años ha crecido la evidencia científica de que los inhibidores del receptor P2Y12 clopidogrel, prasugrel y ticagrelor ejercen efectos beneficiosos más allá de sus efectos antiplaquetarios, los llamados efectos pleiotrópicos56. La explicación potencial es que, aparte de en las plaquetas, el receptor P2Y12 también se detecta en una amplia variedad de tejidos (subregiones del cerebro, células del músculo liso vascular, leucocitos, macrófagos, células dendríticas y células de la microglía), lo que incrementa la disponibilidad de potenciales dianas para el bloqueo del receptor P2Y1257,58. Recientemente, en modelos experimentales se ha demostrado que tanto el clopidogrel como el cangrelor ejercen un efecto beneficioso en el contexto del infarto agudo de miocardio, al reducir la extensión de la lesión cardiaca por mecanismos independientes de su actividad antiplaquetaria59. Estos efectos beneficiosos se han atribuido mayormente a que puede tener un efecto similar al del poscondicionamiento (p. ej., activación de vías de señalización de cardioprotección endógena)60, aunque aún quedan por descifrar los mecanismos que se desencadenan en la activación de esta vía. Un análisis retrospectivo en pacientes con SCA con elevación del ST respalda la capacidad del clopidogrel de atenuar el daño miocárdico por reperfusión61, especialmente cuando se administra en dosis de carga de 600 mg62. Por otro lado, también se ha demostrado que estos fármacos pueden tener efectos pleiotrópicos por mecanismos independientes de su interacción con el receptor P2Y12 (p. ej., incrementando la disponibilidad de adenosina). En este contexto, el ticagrelor ha mostrado un efecto inhibidor en el transportador de nucleósidos equilibrador tipo 1 (ENT-1), presente en los glóbulos rojos. Este bloqueo previene la captación de adenosina por los hematíes, con lo cual aumentan su concentración y su actividad biológica, particularmente en los sitios de isquemia y lesión tisular en los que se libera por falta de oxígeno (Figura 3). La adenosina, a su vez, tiene importantes efectos beneficiosos vasodilatadores, antiinflamatorios y cardioprotectores. Un mecanismo alternativo mediante el cual el ticagrelor puede incrementar la disponibilidad local de la adenosina estaría determinado por su capacidad de incrementar la liberación de ATP por los eritrocitos (Figura 3). Una vez en la circulación, el ATP se degrada a adenosina por las enzimas ecto-ADP-ásicas (CD39 y CD73) presentes en las células endoteliales, las células blancas y los propios glóbulos rojos63. Estudios en pacientes con SCA han demostrado que la administración de ticagrelor incrementa la adenosina circulante, lo que se acompaña de una mejora en el flujo coronario64,65. En contrapartida, sin embargo, el incremento de adenosina puede conllevar efectos adversos como depresión respiratoria o disneas56,66. A pesar de que ensayos clínicos de referencia han puesto de manifiesto que tanto el prasugrel (TRITON-TIMI 38)41 como el ticagrelor (PLATO)51 producen mayor beneficio clínico que el clopidogrel sin aumento concomitante de complicaciones hemorrágicas, estos beneficios han sido especialmente llamativos en el estudio PLATO a favor del ticagrelor. Es cierto que la superioridad de estos nuevos agentes puede derivar exclusivamente de una mejor farmacocinética y de una inhibición plaquetaria más potente, aunque también se puede considerar un posible beneficio derivado de estos efectos pleiotrópicos.

Figura 3. Mecanismos antiplaquetarios y extraplaquetarios (pleiotrópicos) del ticagrelor. Además de prevenir la activación plaquetaria al unirse de manera reversible al receptor P2Y12, también tiene otros efectos beneficiosos en la salud cardiovascular mediados, en gran medida, por su capacidad de incrementar las concentraciones locales de adenosina. El ticagrelor es capaz de prevenir la reabsorción de adenosina mediante el bloqueo de los receptores ENT-1 (transportador de nucleósidos equilibrador tipo 1) y estimular la liberación de trifosfato de adenosina (ATP) por los eritrocitos circulantes, el cual se metaboliza en adenosina por la acción de distintas ecto-ADP-asas.
ConclusionesEn los últimos años ha habido un considerable progreso en el desarrollo de nuevos agentes antiplaquetarios, especialmente los inhibidores del receptor plaquetario P2Y1245. El prasugrel se ha demostrado más potente que el clopidogrel, esencialmente debido a un metabolismo hepático más eficiente, aunque por su irreversibilidad y los riesgos de sangrado, su utilidad está limitada clínicamente a los pacientes definidos en el estudio TRITON. El ticagrelor es un fármaco con gran potencialidad para pacientes con SCA no solo por su efecto directo y reversible en el receptor P2Y12, sino también por sus efectos beneficiosos más allá del antiplaquetario. Si bien sus efectos pleiotrópicos pueden haber contribuido a su impacto clínico, también se han evidenciado efectos adversos (disneas). El cangrelor y el elinogrel, pese a encontrarse aún en desarrollo clínico, ofrecen la posibilidad de un rápido bloqueo reversible del receptor P2Y12 tras su infusión intravenosa. Es más, el elinogrel ofrece la posibilidad de bloqueo del receptor mediante administración tanto oral como parenteral. Sería deseable que se realizaran ensayos multicéntricos de comparación directa entre los nuevos agentes que permitan determinar la seguridad y la eficacia de cada compuesto tanto en SCA como en cirugía coronaria.
FinanciaciónEl trabajo contenido en esta revisión ha sido financiado por el Programa Nacional de Salud (SAF2013-42962-R concedido a LB; SAF2012-40208 concedido a GV).
Conflicto de interesesGV es Investigador Ramón y Cajal con un contrato con el Ministerio de Innovación y Ciencia de España (RyC-2009-5495, MICINN, España).
Agradecimientos
Agradecemos a la Fundación Jesús Serra de Barcelona su continuo apoyo.
Autor para correspondencia: Centro de Investigación Cardiovascular, Sant Antoni M. Claret 167, 08025 Barcelona, España. lbadimon@csic-iccc.org