
Dystroglycanopathies are a heterogeneous group of autosomal recessive disorders with a broad clinical spectrum.1 They include congenital muscular dystrophy, limb girdle muscular dystrophy (LGMC), and dilated cardiomyopathy (DCM). They are characterized by hypoglyocosylation of α-dystroglycan, which is essential for muscle integrity. Fukutin is one of the proteins involved in its glycosylation and in the pathophysiology of the dystroglycanopathies.2 We report the first case described in Europe of a patient with DCM and LGMC who is homozygous for the p.Gly424Ser genetic variant in the fukutin gene (FKTN) and provide histological confirmation of the resulting abnormality.
A 28-year-old man consulted due to limb weakness and myalgia, with no cardiac symptoms. He showed a 4/5 strength loss in the lower limbs, pseudohypertrophy of the triceps surae and gastrocnemius, atrophy of the quadriceps, positive Gower's sign (use of the upper limbs to get up), and elevated creatine kinase (CK; 6220 U/L). Electromyography showed moderate myopathy in all 4 limbs. Electrocardiography revealed sinus rhythm, a short PR interval, and negative lateral Q and T waves, whereas echocardiography showed DCM with 30% left ventricular ejection fraction. After ruling out coronary heart disease, we began treatment with carvedilol and enalapril. The workup was completed with a genetic study of the dystrophin gene, which was normal. Written consent was obtained from the patient for the presentation and publication of this article, including the images. Approval was also received from the institutional ethics committee.
The patient was cardiologically and neurologically stable for 15 years. At 43 years of age, he started showing progressive dyspnea on moderate exertion and was admitted with heart failure and in cardiogenic shock. Electrocardiography (figure 1A) revealed biatrial enlargement, left anterior fascicular block, and lateral Q waves. Echocardiography (figure 1B) showed DCM with severe dysfunction (20% left ventricular ejection fraction) and akinesia in inferolateral segments. He received optimal neurohormone treatment and underwent automatic defibrillator implantation. Due to unfavorable progression, he required a heart transplant 6 months later. In a histological study, the explanted heart showed extensive biventricular fibrosis.
 trends. D: family tree. FKTN: fukutin gene.")
Five years after the transplant, the patient's myopathy was stable but his CK levels were still elevated (figure 1C). He was referred to the inherited heart disease unit, where a family study was performed. His first-degree relatives were healthy and without myopathy, except his father, who had nonobstructive hypertrophic cardiomyopathy (HCM) (maximum septal thickness, 16mm). A genetic study performed using next-generation sequencing with a panel of 18 genes (figure 1D) failed to identify any pathogenic variant associated with HCM.
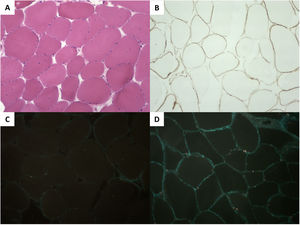
A genetic study performed with next-generation sequencing of the proband (DCM panel, 96 genes) identified the homozygous variant c.1270G>A, p.Gly424Ser in FKTN. His parents were heterozygotic carriers of this variant. A biopsy of the triceps surae muscle revealed mild muscle involvement, with immunohistochemistry confirming a severe deficit in α-dystroglycan and a slight reduction in laminin α2; the remainder of the study was normal (figure 2).
 muscle fibers with slight variability in the diameter and the occasional internalized nuclei. B: (immunohistochemistry) slight reduction in laminin α2. C: severe reduction in α-dystroglycan vs a healthy control (D).")
Histology of the triceps surae muscle. A: (hematoxylin and eosin) muscle fibers with slight variability in the diameter and the occasional internalized nuclei. B: (immunohistochemistry) slight reduction in laminin α2. C: severe reduction in α-dystroglycan vs a healthy control (D).
Fukutin is a ribitol 5-phosphate (Rbo5P) transferase localized to the Golgi apparatus that functions with fukutin-related protein to add Rbo5P pairs to α-dystroglycan, which is mainly expressed in the skeletal muscle, heart, and brain.2 Homozygotic and heterozygotic carriers of FKTN variants show a wide phenotypic spectrum, caused by loss of fukutin function. FKTN was initially described in Japan as a cause of Fukuyama-type congenital muscular dystrophy; it was identified as the founder variant of that country and was also linked to Walker-Warburg syndrome: both conditions comprise a severe phenotype with cerebral involvement and short life expectancy. Cases were subsequently described around the world, as well as milder phenotypes with no cerebral involvement, such as LGMC (type R13). Generally, missense variants cause a milder phenotype, whereas nonmissense variants cause a more severe form, although there are exceptions.3 The clinical symptoms of DCM become apparent from the second decade of life and an infrequent form of DCM with limited or absent muscle involvement has been described.1,4,5 It seems that there is a dissociation between the muscle and cardiac involvement; fukutin has been reported to have other functions and to be essential in the heart to maintain contractility, calcium homeostasis, Golgi apparatus integrity, and mitochondrial stress resistance.6
The p.Gly424Ser variant is localized to a region involved in the phosphorylation of the C-terminal end of FKTN. It appears with low frequency (< 0.01%) in the control population in gnomAD, without homozygotic carriers; the affected residue is highly conserved and replacement of glycine with serine is classified as pathogenic in bioinformatic predictions. It has recently been reported, also in homozygosis, in 2 Mexican siblings with DCM who died at 20 and 21 years of age, with no apparent neuromuscular involvement4; however, no information was provided on CK status or the histological results.
Our case, the first described in Europe, confirms the probable pathogenicity of the p.Gly424Ser variant in FKTN. This variant is associated with mild LGMC that is predominated by cardiac involvement in the form of DCM. This case highlights the importance of genetic studies in patients with DCM and muscle involvement and even in isolated cases, given the possibility of autosomal recessive inheritance.
FUNDINGNo funding has been received to perform this study.
AUTHORS’ CONTRIBUTIONSJ.M. Larrañaga-Moreira and R. Barriales-Villa performed the design, figures, and drafting of the manuscript. P. Blanco-Arias helped to perform the genetic study and critical analysis. B. San Millán-Tejado assisted with the histological study, figure 2, manuscript revision, and critical analysis. G. Barge-Caballero and M.G. Crespo-Leiro helped in the manuscript drafting and critical analysis.
CONFLICTS OF INTERESTP. Blanco-Arias is an employee of Health in Code S.L. The other authors do not declare conflicts of interest.