We read with interest the article by San Román et al.,1 recently published in Revista Española de Cardiología. The results of their registry confirm that COVID-19 is associated with a worse prognosis in heart disease patients, who show a higher incidence of respiratory failure and high mortality rates. The study reports very elevated in-hospital mortality in patients with heart disease (43%), and an even higher rate among those diagnosed with cardiomyopathy (64%).
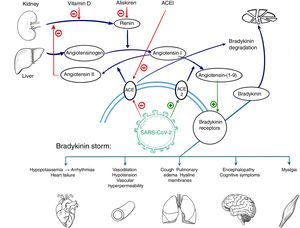
The reasons for the worse prognosis of COVID-19 in patients with previous heart disease has not yet been fully explained. One hypothesis is that it could be related to the mechanisms described by Garvin et al.2 in a study analyzing gene expression in cells from bronchoalveolar lavage material in COVID-19 patients. These authors report a decrease in angiotensin converting enzyme (ACE) expression and an increase in expression of ACE2, an enzyme homologous to ACE that is the entry point of the virus into cells and a mediator of angiotensin I conversion(1-9) and angiotensin II conversion(1-7) into angiotensin. ACE2 overexpression during SARS-CoV-2 infection would lead to increased angiotensin production,(1-9) which has a sensitizing effect on receptors of bradykinin, a vasodilatory peptide that is degraded by ACE under normal conditions (figure 1). Thus, in SARS-CoV-2 infection, bradykinin would have a more potent and persistent action, a situation known as “bradykinin storm”, in which vascular dilation and permeability would be increased (among other effects), thereby triggering many of the symptoms related to a poor clinical course during COVID-19.

Patients with previous heart disease could be more vulnerable to this pathophysiological mechanism due to higher baseline production and release of renin. Among other events associated with elevated renin, which is the limiting factor of angiotensinogen conversion to angiotensin I, it has been related to a higher prevalence of heart failure and sympathetic hyperactivation.3 Durante SARS-CoV-2 infection, ACE2 overexpression would convert excess angiotensin I into angiotensin-(1-9) and this would lead to a more marked effect of bradykinin, with consequent clinical worsening. The effect of ACE inhibitors is controversial in this situation. Although they cause a renin increase by decreasing angiotensin II production, it is unknown whether this effect is added to effect produced by SARS-CoV-2 by this same mechanism. It is possible that chronic administration of these agents could induce mechanisms alternative to ACE to inactivate bradykinin,4 which might be beneficial during COVID-19.
The promising results of the recent pilot clinical trial by Trainas Castillo et al.,5 in which calcifediol (25-hydroxyvitamin D) administration was associated with a significant reduction in intensive care unit admissions in COVID-19 patients, support the hypothesis that bradykinin storm could be a trigger for respiratory failure in this disease. Vitamin D is an inhibitor of renin production,6 which would limit angiotensin I production. Another potentially beneficial drug, according to this hypothesis, would be aliskiren, a direct, selective renin inhibitor. It would be of interest to research the therapeutic effect of this agent against COVID-19 in clinical trials.