Cardiac amyloidosis is a disease of complex diagnosis and treatment. Some subtypes of cardiac amyloidosis are inherited. Among these, the most common variant is caused by mutations in the transthyretin gene.
Correct identification of amyloidosis produced by a genetic defect is of great importance because it modifies the diagnostic and therapeutic approach in patients and their families.
We describe our experience in the evaluation of two families with hereditary transthyretin cardiac amyloidosis. We discuss a number of considerations related to the evaluation of these patients and the diagnostic and therapeutic approach to perform in relatives.
Keywords
Cardiac amyloidosis is a disease caused by the deposition of an insoluble proteinaceous material known as amyloid.1, 2 The different possible origins and compositions of this abnormal material are responsible for the different subtypes of amyloidosis, each one with a different prognosis and specific treatment.1, 2
Among the different subtypes of amyloidosis are hereditary forms, in which the deposition arises due to a genetic abnormality. Of the different hereditary forms of amyloidosis, only the apolipoprotein A-I (AL) and transthyretin (TTR) forms are involved in cardiac amyloidosis. The latter of these is much more common.1
TTR is a protein synthesized in the liver. More than 100 mutations have been described in the gene that encodes the protein. The different mutations can give rise to different phenotypes, and neuropathic, cardiac, renal, and ocular forms have been reported.1, 2
Given that many heart diseases are hereditary in nature and that genetic testing is now possible, cardiologists should investigate such possible diagnoses in their clinical practice. Appropriate interpretation of genetic results will be of importance for genetic counseling and decision making in the diagnosis, follow-up, and treatment of patients and their relatives.
MethodsWe describe our experience in the diagnosis and assessment of 2 families with hereditary TTR cardiac amyloidosis.
Results Family 1 Index PatientThe index patient was a 52-year-old man with a history of arterial hypertension, hypercholesterolemia, and bilateral carpal tunnel syndrome (CTS) for which he had undergone surgery.3 When he was 48 years old, he required a pacemaker for atrial fibrillation and slow ventricular response. Two years later, an echocardiogram was recorded after signs and symptoms of heart failure. The study revealed restrictive cardiomyopathy consistent with a deposition disease. The endomyocardial biopsy revealed hyaline deposits positive for Congo red.
Despite the normal findings in blood and urine electrophoresis and immunofixation and normal bone marrow biopsy, AL amyloidosis was diagnosed, and a watch-and-wait approach was decided.
During the next 2 years, the patient experienced progressive deterioration in his functional capacity, with frequent admissions for heart failure and decreased ejection fraction. He was therefore referred for consideration for heart transplantation (HT).
The clinical presentation, which was not typical of AL amyloidosis due to its slow progression, the absence of a monoclonal peak in blood and urine, and a normal bone marrow biopsy lead to suspicion of an alternate diagnosis. 99mTc-3,3-diphosphono-1,2-propanodicarboxylic (99mTc-DPD) scintigraphy showed myocardial uptake, and immunohistochemical study of the cardiac sample revealed TTR deposits (Figure 1). The genetic study identified a heterozygous Glu89Lys mutation in the TTR gene, as described previously.4 Electroneurography (ENG) showed evidence of mild-moderate sensorimotor polyneuropathy.

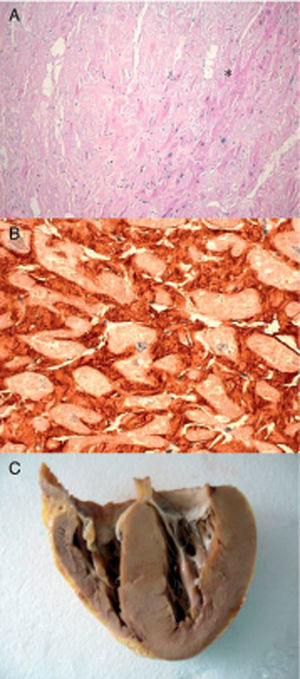
Figure 1. Endomyocardial biopsy of the patient with hereditary transthyretin cardiac amyloidosis. A: Hematoxylin-eosin. Amyloid (*) appears as amorphous material between myocytes. B: Immunohistochemistry for detecting transthyretin around the myocytes. C: Heart of index patient 2. Courtesy of Dr. Clara Salas Antón.
In view of the clinical worsening and the absence of significant involvement of other body systems, HT was proposed, and this was performed without any complications. Six months later, the patient was put on a waiting list for liver transplantation (LT). He waited for 18 months, during which time amyloid was not detected in the endomyocardial biopsy and there was no significant progression of the neuropathy. Four years after HT and 2 years after LT, the patient is leading a normal life.
Study of Family MembersThere was no family history of heart disease or sudden death. His mother had died at age 51 from tuberculosis and his father was still alive at age 72, with no significant conditions.
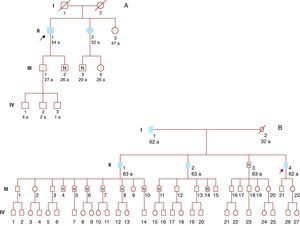
A genetic study was offered to relatives over 16 years of age. Six family members were studied, and 4 were found to be carriers of the same genetic mutation (Figure 2A).

Figure 2. A: Family 1. B: Family 2. Filled symbols: Affected individuals. White symbols: Without heart disease. Symbols with dot: Carriers without heart disease. Symbols with N: Noncarriers. Symbols with strike-through: Dead. Arrow: Index patient.
All carriers were recommended to attend annual follow-up and undergo regular cardiac imaging and ENG.
Our hospital only follows subject III:1 who, after almost 5 years, has not presented with any clinical signs of neuropathy or heart disease. In the most recent visit, no amyloid deposits were detected by cardiac magnetic resonance imaging (MRI) or neuropathic disease by ENG.
The patient was given the appropriate genetic counseling. His 3 children have not undergone genetic study as they are still minors.
Of the remaining relatives who were carriers of the mutation, we are aware that one of the sisters of the index patient, with a history of CTS, has experienced heart failure at 52 years of age. Despite our recommendations, she declined the offer to be considered for HT and LT.
Family 2 Index PatientThe index patient was a 62-year-old man with a history of arterial hypertension, diabetes mellitus, and bilateral CTS. He presented with symptoms of heart failure, which were interpreted as being caused by hypertensive heart disease, until an echocardiogram showed signs of restrictive cardiomyopathy of infiltrative appearance. A rectal biopsy was performed and revealed deposits positive for Congo. By means of immunohistochemistry, these deposits were identified as TTR.
ENG showed evidence of moderate sensorimotor polyneuropathy. After a deterioration in functional class, the patient was referred for assessment.
The prognostic study of the patient's heart disease showed unfavorable parameters, and right heart catheterization showed highly elevated pulmonary artery pressures and a reduced cardiac output. In the MRI, gadolinium uptake was observed throughout the myocardial walls. 99mTc-DPD scintigraphy showed cardiac uptake.
A genetic study revealed an already described heterozygous mutation consistent with deletion of codon V122 of the TTR gene.
In view of the elevated pulmonary resistances contraindicating HT, it was decided to treat the patient with a combination of sildenafil and bosentan. After 3 months of treatment, which was well tolerated, right heart catheterization was performed once again, and showed reduced pulmonary resistance. Given the advanced functional class and the need for frequent admissions, HT followed by LT was considered once again.
The HT was associated with early right ventricular failure that required implantation of right ventricular assistance. After progressive improvement and withdrawal of ventricular assistance (28 days after HT), the patient experienced numerous infectious complications associated with prolonged stay in the intensive care unit and he died 50 days after the procedure.
Study of Family MembersThe father of the subject had died at age 62 from heart failure. Two of his brothers had died at 63 years of age after progressive heart failure (one due to sudden death). In subject II:2, diagnosis of amyloidosis was made, although the subtype was not identified.
Sixteen relatives underwent genetic study and 9 had the mutation (Figure 2B). The only living brother of the index patient was not a carrier of the mutation, and so it was not necessary to study his descendants. Several descendants of his dead brothers had the mutation, and therefore were obligate carriers.
Our hospital follows 3 of the carrier relatives (III:2, III:3, III:21). They are clinically asymptomatic and their cardiac examinations are normal. Unlike his family members and despite being asymptomatic, subject III:2, aged 45 years, showed signs of moderate multiple neuropathic involvement in ENG and signs compatible with right CTS.
All carriers of the mutation were offered genetic counseling and we are monitoring several at a distance to determine the best opportunity for LT.
DiscussionIdentification of patients whose amyloidosis is of genetic origin is of utmost importance, as it affects treatment decisions and is also important for relatives.1, 2, 5
Hereditary causes should always be considered in these patients, as up to 10% of the subjects diagnosed with AL amyloidosis may have hereditary amyloidosis.5
In our experience, use of immunohistochemical techniques facilitates the appropriate identification of the subtype of amyloidosis in the biopsy (which does not necessarily have to be a cardiac biopsy) and avoids erroneous diagnoses and delays in initiating therapy, as was the case in the first patient.
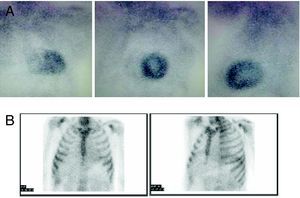
To facilitate diagnosis of the subtype of cardiac amyloidosis, 99mTc-DPD scintigraphy has been suggested as a technique able to distinguish between hearts with TTR amyloid (with uptake) and AL amyloid (without uptake).6 Our initial experience with this technique in the 2 patients presented and a further 4 with AL amyloidosis concurs with what has been previously published (Figure 3).

Figure 3. 99mTc-DPD scintigraphy. A: Hereditary transthyretin cardiac amyloidosis. B: AL amyloidosis. Note the difference in cardiac uptake. Courtesy of Dr. Javier de Haro del Moral.
Cardiac MRI provides excellent morphologic characterization, with the advantage of allowing study by means of late gadolinium enhancement. The typical gadolinium uptake pattern has been described as global and subendocardial, but localized transmural uptake has also been reported.7 Unfortunately, unlike scintigraphy, MRI cannot differentiate between the different subtypes of amyloidosis.
Once the diagnosis of hereditary amyloidosis has been established and the mutation identified, genetic study of relatives can determine who needs to be monitored and who does not. As noncarrier descendants will not be affected, the number of family members for whom follow-up is necessary decreases.
On the other hand, in carrier subjects, effective genetic counseling—ranging from family planning to career planning—can be given. For example, subject III:3 (family 2) works as a civilian pilot and, in his case, close monitoring of possible clinical neuropathy would affect his professional activity.
We do not recommend genetic studies in those under 16 years of age, given that the result would not affect the clinical approach while having possible negative psychological consequences for the minors and those around them.
Care of patients with amyloidosis requires multidisciplinary units with experience in complex treatments and the capacity for providing genetic counseling.3 In hereditary amyloidoses, the only effective procedure for treating the presence of amyloid is LT.1, 2, 3, 8
Although the phenomenon of anticipation (appearance at increasingly earlier ages in successive generations) has been described in neuropathic forms with a Val30Met mutation, in the cardiac forms the clinical signs and symptoms often do not appear until subjects are in their 40s.
The follow-up we recommend for adult carriers includes clinical check-ups and annual cardiac MRI and ENG every 2 years.
Given that a family member already has preclinical neuropathic disorders, there is debate about when to perform LT to prevent irreversible cardiac involvement. The international expert opinion is that, given the risks, LT should be performed in the first year after the onset of clinical signs and symptoms (Rapezzi, private communication).8
Given that the liver of these patients is otherwise healthy, it can be used in a transplant to suboptimal recipients (“domino” transplant). Unfortunately, a domino transplant may cause amyloidosis in the recipient.
When there is substantial cardiac involvement, the only alternative is a combined heart-liver transplant, as was considered in our patients.
Although it has been suggested that mutated cardiac amyloid deposits might act to “capture” native protein,9 we believe that there is not enough evidence for this to justify a combined transplantation, which is obviously associated with significant risk while almost the only benefit is logistical.
In conclusion, hereditary cardiac amyloidosis is difficult to diagnose and its treatment is complex. Nevertheless, accurate diagnosis is necessary given the large impact of the disease on the outlook of the patients and their families.
Our experience shows the utility of performing a genetic study of family members with the goal of early diagnosis, close follow-up, and early treatment.
FundingWork partially funded by the FIS (PI08/979) and the thematic network for research in heart failure REDINSCOR (RD06/003/0018), Instituto de Investigación Carlos III, Ministerio de Sanidad, Política Social e Igualdad. Patricia Avellana is funded as a fellow of the Carolina-BBVA Foundation.
Conflicts of interestNone declared.
Received 7 June 2010
Accepted 12 October 2010
Corresponding author: Unidad de Miocardiopatías, Sección de Trasplante Cardiaco, Servicio de Cardiología, Hospital Universitario Puerta de Hierro, Manuel de Falla 2, 28222 Majadahonda, Madrid, Spain. pablogpavia@yahoo.es