Genetics has rightly acquired an important place in almost all medical disciplines in recent years and this is certainly the case in the field of congenital cardiology. Not only has this led to greater insight into the pathophysiology of congenital heart defects but it also has a beneficial impact on patient management. Integration of clinical genetics in multidisciplinary centers of expertise for CHD is therefore a clear recommendation. Adult and pediatric cardiologists play a crucial role in the process of genetic evaluation of patients and families and should have be familiar with red flags for referral for further clinical genetic elaboration, counseling, and eventual testing. Some basic knowledge is also important for the correct interpretation of genetic testing results. In this review article, we provide a practical overview of what genetic evaluation entails, which type of genetic tests are possible today, and how this can be used in practice for the individual patient.
Keywords
Congenital heart defects (CHD) are among the most common birth defects, affecting approximately 1% of live born children.1 Significant progress has been made in recent years in both the accuracy of clinical diagnosis and the treatment of CHD. This has increased survival and enhanced quality of life in a large proportion of affected patients, with adults with CHD now outnumbering children in many countries.2 Along with this progress, the demand and search for possible explanations of the underlying cause of the heart defect has grown in recent years, both from patients and parents, as well as from the care providers involved. Recent technological advances spearheaded our understanding of the genetic basis of syndromic forms of CHD. In isolated CHD, however, knowledge of the molecular mechanisms is largely absent, although several lines of evidence indicate that genetics has an important contribution to make: the incidence of CHD affecting both twins is higher in monozygotic than in dizygotic twins,3 the recurrence risk of CHD for siblings and offspring of patients with CHD is higher than in the general population,4,5 and using high throughput technologies allows identification of a genetic anomaly in up to one third of patients with CHD, taking syndromic and nonsyndromic forms together.6
Knowledge of the underlying genetic cause(s) will help to fine tune personalized counseling and treatment options. It has already been established for various lesions that an underlying genetic defect influences the management and outcome of CHD. For example, patients with an atrial septal defect (ASD) due to NKX2.5 pathogenic variants should be carefully monitored for arrhythmias.7 Furthermore, especially in children, the underlying defect may indicate a risk for noncardiac complications such as neurodevelopmental delay, respiratory problems or renal dysfunction, requiring early intervention or follow-up to prevent or alleviate these manifestations.
Finally, knowledge of the genetic basis will affect counseling for the recurrence risk in siblings and offspring and may provide access to reproductive options using prenatal and preimplantation genetic diagnosis. Over the past decades, integration of epidemiological, clinical and genetic data have improved knowledge of CHD recurrence significantly. The prevailing hypothesis of CHD being inherited as a multifactorial trait was already challenged during the 1980s.8 Rose et al.9 observed a higher than expected occurrence of CHD based on multifactorial models in several families. Indeed, increased use and further refinement of imaging techniques led to the important observation that some (but not all) lesions belong to a broader phenotypic CHD spectrum that may occur in a familial context. Established examples are left-sided outflow defects: family members of children with hypoplastic left heart syndrome or severe left ventricular outflow tract (LVOT) obstruction were found to have a bicuspid aortic valve (BAV) at higher than expected rates.10,11 In addition to the above, knowing the underlying cause may have a “therapeutic” effect, helping patients and their relatives to cope with and accept a rare disease.
Novel genetic techniques based on high throughput analyses, shorter turnaround times and at affordable costs have increased accessibility to genetic diagnosis. It is expected that this trend will continue at a rapid pace and genetics will provide answers in a growing number of cases.
Nevertheless, thorough genetic analysis comes with increasing challenges to interpretation of the results, and an ever-changing field of possibilities and drawbacks. This situation has prompted this review on the current status of genetic testing in the field of CHD.
BASICS OF GENETICSDefining geneticsNo testing without counseling!When defining “genetics”, it is important to make a distinction between the concepts of “genetic counseling” and “genetic testing”. We emphasize from the outset that both concepts are inextricably linked—genetic testing must always be accompanied by correct counseling—but counseling will not result in testing in all cases.
According to the World Health Organization, genetic counseling is defined as “the process through which knowledge about the genetic aspects of illnesses is shared by trained professionals with those who are at an increased risk of either having a heritable disorder or of passing it on to their unborn offspring”.
Genetic counseling in the setting of CHD was introduced more than half a century ago, whereby the most important setting was to inform parents of an affected child about their recurrence risk. Early studies already nicely demonstrated that informing parents in a dedicated counseling process had a beneficial effect.12 More recent research has confirmed the beneficial effects of individualized genetic counseling sessions to the parents of children with CHD with regard to improving knowledge about the causes of CHD and enhancing psychosocial functioning, strongly recommending their inclusion in routine clinical practice.13 With increasing numbers of adults with CHD, the indications for genetic counseling and testing have been expanded and “genetics” are listed as a requirement for adult CHD programs in the recently published American College of Cardiology/American Heart Association guidelines on adult CHD.14
Genetic counselors in CHD are graduate level trained health care professionals who have received training in both medical genetics and counseling with a particular focus on CHD. Genetic counselors will draw a 3-generation pedigree and collect all relevant clinical data from the proband and family members with special attention on miscarriages or neonatal deaths. Apart from their role in family history taking and in counseling patients and families about recurrence risk, risk for a specific syndrome and interpretation of results, genetic counselors may play an important role in triaging patients who should be referred for a complete genetic evaluation.15
Genetic elaboration of CHD requires a multidisciplinary approach in which, in addition to the (pediatric) cardiologist and genetic counselor, the clinical geneticist also plays a crucial role. Clinical geneticists are physicians who have undergone specific training in diagnostic evaluation, management, and genetic counseling. Training programs and certification are nation specific. Clinical geneticists will determine whether the heart defect is isolated or part of a syndrome, which is required to guide genetic testing and to determine the medical approach. Based on large epidemiological studies, syndromic cardiovascular malformations comprise at least 25% of all cardiovascular malformations.4,16 Research in the setting of 22q11 deletion has already shown that cardiologists are less good at assessing syndromes and that clinical genetic evaluation is therefore desirable.17 Once it has been determined whether or not a patient has a syndromic entity, the medical management of syndromic forms can also be better coordinated by a clinical geneticist in the context of a multidisciplinary team —patients with isolated CHD forms are of course best followed up by the (pediatric) cardiologist.8
Another important issue to take into account in the process of genetic counseling and testing is consent. It is essential for any genetic test that the patient (or his or her legal representative) is aware of the benefits and risks of such testing and gives written consent for the test. It is outside the scope of this article to discuss (important) aspects such as incidental findings and presymptomatic testing, but we would like to briefly address direct-to-consumer (DTC) testing.
In many countries, long gone are the days when genetic testing was confined to certified clinical genetic centers (laboratory-directed testing) for which strict rules apply for conducting clinical and molecular diagnostics. As a result of the technical progress in genetic testing on the one hand, and the increasing public demand on the other hand, significant growth has been observed in companies that offer testing known as DTC. These are tests in which samples (blood or saliva) are directly mailed to the laboratory, without preemptive counseling. DTC genetic tests may detect severe and highly penetrant monogenic disorders or genetic variants associated with increased susceptibility for common and complex diseases. There are concerns that variant interpretation from DTC testing may not always be correct. There have already been reports of cases of unnecessary treatment in healthy family members or false reassurance based on incorrect information.18 One study showed that 40% of variants in a variety of genes reported in DTC raw data were false positives.19 This false information severely impacts patients and families in the first place but also overloads genetic counseling services who are consulted to clarify and rectify the results of tests ordered elsewhere.20 It goes without saying that these issues create tension in the context of DTC genetic testing regarding the expectations and normative assessment of communication strategies.21
For these reasons, the European Society of Human Genetics has developed a policy on the advertising and provision of predictive genetic tests by such DTC companies.22 We argue against the use of DTC testing in the CHD genetic testing context.
The technical (r)evolution of genetic testingEvolution in cytogeneticsThe seminal discovery of trisomy 21 as the genetic cause of Down syndrome in 195923 introduced genetic testing in CHD. Since then, novel methodologies and technical fine tuning have been instrumental in identifying the genetic cause of (mainly syndromic forms) of CHD. Classic karyotyping with G-banding has a rather low resolution of 3-5 Mb, and is currently only performed for specific indications, such as the confirmation of (mozaic) aneuploidies (Down syndrome, Turner syndrome, mosaic trisomy 8) and familial translocations.
Fluorescent in situ hybridization (FISH) makes use of a fluorescently labeled probe targeting specific genomic regions. It is used for the targeted detection of aberrations below karyotyping resolution, such as the 22q11.2 or 7q11.2 microdeletions in velocardiofacial syndrome and Williams-Beuren syndrome, respectively. A major breakthrough came with the introduction of array comparative genome hybridization (ArrayCGH),24 also called chromosomal microarray analysis. Chromosomal microarray analysis competitively hybridizes shredded DNA of control and patient DNA, labeled with different fluorochromes on an array containing tens of thousands of molecular probes dispersed over the reference genome. Next, automatic reading of differences in color intensities detects genome-wide copy number variations (CNVs), ie, deletions or duplications, as small as 100kb This technique is also referred to as “molecular karyotyping”. Single nucleotide polymorphism (SNP) array is a similar test that uses SNPs to detect regions with a loss of heterozygosity. However, the huge amount of structural variability in the human genome has hampered straightforward interpretation of test results, especially in the years following the introduction of the test in the diagnostic setting. Initiatives such as the Database of Genomic Variants25 have been detrimental in documenting normal variation while databases such as DECIPHER26 have played a prominent role in identifying novel genomic structural defects as the basis of disease. Indeed, molecular karyotyping introduced the concept of “reversed genetics”, a strategy whereby patients with the same genetic variant are compared to identify a genotype-phenotype correlation and delineate novel clinical entities, such as Koolen-de Vries syndrome.27 The most frequently associated structural variants in CHD known to date are reported in more detail below. More recently, chromosomal microarray analysis is being replaced by methods based on low-coverage (“shallow”) genome sequencing technologies (see below).
Evolution in molecular geneticsThe combined use of polymerase chain reaction and Sanger sequencing introduced the use of molecular analysis in the clinic. Nevertheless, analysis was expensive and time-consuming. Moreover, the identification of novel genes causing cardiovascular phenotypes was restricted to syndromic forms that could be investigated through linkage analysis in large families with dominant inheritance for the condition (eg, Noonan syndrome28), or in consanguineous families with recessive conditions (eg, Ellis-van Creveld29), while candidate gene approaches only sporadically identified a casual defect, often helped by the previous detection of a microdeletion encompassing the candidate gene (eg, CHARGE syndrome30). A second breakthrough came with the introduction of next-generation (or massively parallel) sequencing (NGS).31 In brief, in NGS, DNA fragments of the region(s) of interest (either a specific panel of genes, the exome, ie, the coding regions of the DNA, or the genome, ie, the whole DNA sequence) are sequenced in parallel and the obtained “reads” are aligned to the reference sequence. The coverage at a certain genomic position refers to the number of times a base at a certain genomic position is independently sequenced. For a reliable interpretation, at least a coverage of 20x is necessary. Short read sequencing methods are primarily used in clinical laboratories because of their cost-effectiveness and low per-base error rate. The application of exome sequencing may than help to either analyze an extensive number of genes known to cause CHD and even to identify novel CHD candidate genes. However, short read lengths (50–500bp) can produce misalignments and misassemblies in areas of high genome complexity, are unable to cover repeats reliably, and impair phasing of variants. Furthermore, the amplification process, which is indispensable in short read sequencing, creates an underrepresentation of bases in areas of high or low guanine-cytosine (GC) content.
Again, due to the huge variability in the human genome, variant interpretation is crucial, based on freely accessible databases and causality prediction with bioinformatic tools (see below). Similar to DECIPHER, databases such as GeneMatcher32 arose to catalyze rare disease discovery by providing a platform to connect clinicians and researchers from around the world who share an interest in the same gene.
Third generation sequencing: evolution to a single genomic test?Despite copy number analysis (with a resolution of at least 100kb) and high performant sequence analysis using short read NGS, most of the structural variation is still missed. Structural variants comprising CNVs, inversions, and translocations make up to 10% of the genome and contribute greater diversity between 2 human genomes than any other form of genetic variation and may affect expression of genes.
In addition to large chromosomal defects such as translocations, inversions and especially CNVs, smaller cryptic structural variants (ranging from 50bp to 50kb) can also cause human diseases by affecting gene function or expression; eg, structural variants >20 kbp are up to 50-fold more likely to affect the expression of a gene compared with an SNV.
Third generation genome sequencing platforms use long read sequencing (LRS, >10kb reads) and enable the elucidation of structural variations at a previously unparalleled resolution and overcome most of the shortcomings of short read sequencing.33 This will eventually lead to a single analysis to cover most genomic DNA variation within the near future.
Third generation genome sequencing can be combined with other NGS approaches, such as transcriptome sequencing (library of expressed genes in a certain cell type) to identify variation at the level of expression and splicing. Nevertheless, the main hurdle will remain the interpretation of the variants, which often requires additional validation and which will remain the main burden in the diagnostic application of these techniques (see below).
Interpretation of genetic test resultsGene curation needWith the technical progress of genetic testing as described above, now enabling simultaneous testing of large numbers of genes, the temptation has been great to effectively set up more extensive panels for specific disorders. Commercial genetic laboratories in particular have taken this path and now offer panels for CHD that contain> 100 genes, for example.
However, some caution is advised in this trend. For multiple disorders, there is strong evidence that testing more genes inevitably leads to detection of more “variants of unknown significance” or VUSs, the interpretation of which is not easy and sometimes even risks creating unnecessary anxiety and discrimination in patients.34,35 When selecting genes for inclusion in diagnostic panels or reporting from exome/genome sequencing, the clinical validity, ie, the strength of evidence that variation in that gene predisposes to the disease, needs to be carefully considered. A framework for semiquantitative assessment of gene-disease validity has been developed for many diseases (but not yet for CHD) by the Clinical Genome Resource, or ClinGen. In this framework, genes are classified into prespecified tiers based on the clinical, genetic and experimental evidence, along with discussion and consensus of clinical domain experts. These clinically validated genes can be used to prioritize genes for research and inform which genes should be included in disease panels.36,37
Variant curation needThe possibility of generating large amounts of genetic data through broad genetic screening has allowed rapid and more accurate genetic diagnosis. However, as already mentioned, each analysis yields multiple genetic variants (up to 50 000 variants per exome), which need appropriate interpretation. Distinguishing benign from pathogenic variants is essential for translating genetic results into clinical practice but remains challenging. Moreover, the number of VUSs is still too high. In 2015 the American College of Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) proposed a guideline to classify genetic variants for Mendelian disorders.38 Classification is based on 28 criteria to finally classify a variant as benign (class 1), likely benign (class 2), variant of unknown significance (class 3), likely pathogenic (class 4) or pathogenic (class 5), where classes 2 and 4 provide greater than 90% certainty of a variant either being benign or disease-causing. The criteria include clinical findings, data retrieved from large human exome databases such as gnomAD and ExAC and data addressing the structural effect of a variant on the DNA/protein level.
Correct and detailed clinical findings are required, not only of the proband, but often also of family members. Cooperation with—and from—family members is therefore highly important, both in a diagnostic setting and in the further communication of the results. Further segregation of identified variants (both copy number variants and single nucleotide variants) requires verification in first-degree relatives, in which, in addition to DNA studies, clinical cardiovascular examination is also required to correctly assess the status of the individual. It is important to communicate this properly to the patient from the beginning of the counseling and testing process.
If an abnormal test result is confirmed, it is also recommended to check the result further with additional family members. Caregivers are not permitted by law to contact family members; contact must go through the patient (or his or her representative) and it is also important to communicate this correctly to the patient when discussing the results.
In addition to clinical criteria in patients and family members, the molecular characteristics of the variants are also taken into account. Computational analysis of a variant with modeling of the expected effects of the gene or variant on protein structures and function can provide supporting evidence to establish pathogenicity. Despite careful interpretation and international initiatives for data curation, the clinical consequences for many variants obtained through in-depth molecular analysis remain unknown. Several tools, including transcriptome, proteomic, metabolomic, lipidomic and methylomic analysis, may help to identify the molecular consequences. However, the respective (multi)omic signatures are unknown for most genes defective in CHD. In addition, some genes may only be relevant during cardiac development and postnatal testing on other tissues might be irrelevant. Also, animal modeling for specific diseases is time-consuming, expensive, and impossible in the clinical setting. However, direct mutagenesis using the CRISPR_Cas9 techniques is becoming increasingly efficient and may eventually be helpful in the interpretation of genomic variants.
Although the ACMG/AMP guidelines definitely introduced major improvements in the interpretation of genetic variants, they often still leave room for subjective interpretation and therefore several groups have proposed more gene specific classifications.39
Since the publication of the guidelines, several tools have been developed to aid the interpretation of genetic variants (table 1). Moreover, several online repositories are also available to consult variants which have been previously classified by other laboratories (table 1). Caution, however, should be exercised when consulting these repositories since variant interpretation remains subjective and is not always performed by experienced groups. Clinvar, one of these archives, not only provides an interpretation of a specific variant, it also provides a level of review supporting the assertion of clinical significance.
Overview of freely available online tools for classification and interpretation of genetic variants
| Tools for the classification of genetic variants | |
|---|---|
| Tool | Description |
| Clingen Pathogenicity Calculator40,41 | Based on ACMG/AMP guidelines and further expert inputNeed for manual data entryRegistration neededOption to directly submit to Clinvar |
| Varsome42,43 | Based on ACMG/AMP guidelines, no expert input? but extra bioinformatic supportAutomated variant interpretationOption of manual modification of the classification |
| Intervar44,45 | Based on ACMG/AMP guidelines, no expert input?Automated variant interpretationOption of manual modification of the classification |
| Franklin46 | Based on ACMG/AMP guidelines, no expert input?Automated variant interpretationOption of manual modification of the classification |
| Cardioclassifier47,48 | Only for cardiovascular diseasesBased on ACMG/AMP guidelines and some specific expert knowledgeAutomated variant interpretation, no option of manual modification of the classificationFree registration needed |
| Online repositories | |
|---|---|
| Clinvar49,50 | Partner of ClinGenClassification is reviewed by experts and assigned a reviewed statusRegular update |
| Leiden Open Variation Database51,52 | Partner in the Human Variome ProjectRegular update of classified variants |
| Universal Mutation Database53,54 | Partner in the Human Genome Variation SocietyData restricted to certain locus-specific variants |
| Human Gene Mutation Database55,56 | Free registration neededRepository of main published data on a certain variant rather than an interpretation archiveRegular update |
ACMG/AMP, American College for Medical Genetics/Association for Molecular Pathology.
Genetics is a dynamic and rapidly evolving field. For many clinical phenotypes, new genomic data are regularly discovered and test findings issued today may be outdated tomorrow. Therefore, regular and careful reconsideration of genetic counseling and testing should take place, especially in those individuals/families with a high level of suspicion but without identification of a genetic defect. In the same line, genetic variant interpretation can change over time and previously found genetic variants (especially VUSs) should be regularly reassessed in light of new published data.57
Again, and even more so than in the initial counseling process, retesting and communication of altered test results should be undertaken with care to ensure that the results are correctly interpreted by patients and their families.58
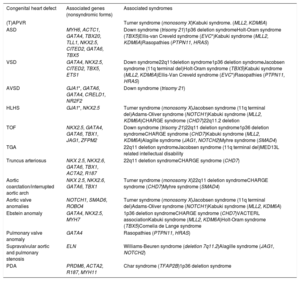
THE SPECTRUM OF GENETIC DEFECTS IN CHDUp to 25% to 30% of patients with CHD have other associated extracardiac manifestations.59 The association of CHD in several chromosomal aneuploidies and CNVs such as Down syndrome, Turner syndrome and 22q11 deletion syndrome has been well-stablished. Other CNVs and single gene variations have also shown high penetrance of CHD. For the other nonsyndromic CHD, several genes showing Mendelian inheritance (mostly autosomal dominant but in some cases also autosomal recessive) have been identified. Of note, some of these genes might be involved in both syndromic and nonsyndromic cases. Table 2 and table 3 provide a summary of several forms of CHD associated with genetic disorders, as well as the most relevant clinical manifestations of the most frequent syndromes. Overall, many genes involve transcription factors, signaling pathways, or chromatin remodelers. Hence, the dosage and alteration of gene expression is a likely relevant mechanism in CHD. Therefore, other mechanisms, including structural variants, might currently be underdiagnosed in CHD. Also, altered gene dosage at critical developmental stages offers a window for environmental factors to interfere with cardiac development. Finally, somatic mosaicism in cardiac progenitor cells is still under debate.
Overview of the different genes and syndromes associated with congenital heart defects
| Congenital heart defect | Associated genes (nonsyndromic forms) | Associated syndromes |
|---|---|---|
| (T)APVR | Turner syndrome (monosomy X)Kabuki syndrome. (MLL2, KDM6A) | |
| ASD | MYH6, ACTC1, GATA4, TBX20, TLL1, NKX2.5, CITED2, GATA6, TBX5 | Down syndrome (trisomy 21)1p36 deletion syndromeHolt-Oram syndrome (TBX5)Ellis-van Creveld syndrome (EVC*)Kabuki syndrome (MLL2, KDM6A)Rasopathies (PTPN11, HRAS) |
| VSD | GATA4, NKX2.5, CITED2, TBX5, ETS1 | Down syndrome22q11deletion syndrome1p36 deletion syndromeJacobsen syndrome (11q terminal del)Holt-Oram syndrome (TBX5)Kabuki syndrome (MLL2, KDM6A)Ellis-Van Creveld syndrome (EVC*)Rasopathies (PTPN11, HRAS) |
| AVSD | GJA1*, GATA6, GATA4, CRELD1, NR2F2 | Down syndrome (trisomy 21) |
| HLHS | GJA1*, NKX2.5 | Turner syndrome (monosomy X)Jacobsen syndrome (11q terminal del)Adams-Oliver syndrome (NOTCH1)Kabuki syndrome (MLL2, KDM6A)CHARGE syndrome (CHD7)22q11.2 deletion |
| TOF | NKX2.5, GATA4, GATA6, TBX1, JAG1, ZFPM2 | Down syndrome (trisomy 21)22q11 deletion syndrome1p36 deletion syndromeCHARGE syndrome (CHD7)Kabuki syndrome (MLL2, KDM6A)Alagille syndrome (JAG1, NOTCH2)Myhre syndrome (SMAD4) |
| TGA | 22q11 deletion syndromeJacobsen syndrome (11q terminal del)MED13L related intellectual disability | |
| Truncus arteriosus | NKX 2.5, NKX2.6, GATA6, TBX1, ACTA2, R187 | 22q11 deletion syndromeCHARGE syndrome (CHD7) |
| Aortic coarctation/interrupted aortic arch | NKX 2.5, NKX2.6, GATA6, TBX1 | Turner syndrome (monosomy X)22q11 deletion syndromeCHARGE syndrome (CHD7)Myhre syndrome (SMAD4) |
| Aortic valve anomalies | NOTCH1, SMAD6, ROBO4 | Turner syndrome (monosomy X)Jacobsen syndrome (11q terminal del)Adams-Oliver syndrome (NOTCH1)Kabuki syndrome (MLL2, KDM6A) |
| Ebstein anomaly | GATA4, NKX2.5, MYH7 | 1p36 deletion syndromeCHARGE syndrome (CHD7)VACTERL associationKabuki syndrome (MLL2, KDM6A)Holt-Oram syndrome (TBX5)Cornelia de Lange syndrome |
| Pulmonary valve anomaly | GATA4 | Rasopathies (PTPN11, HRAS) |
| Supravalvular aortic and pulmonary stenosis | ELN | Williams-Beuren syndrome (deletion 7q11.2)Alagille syndrome (JAG1, NOTCH2) |
| PDA | PRDM6, ACTA2, R187, MYH11 | Char syndrome (TFAP2B)1p36 deletion syndrome |
ASD, atrial septal defect, AVSD, atrioventricular septal defect; HLHS, hypoplastic left heart syndrome; PDA, persistent ductus arteriosus; (T)APVR, (total) abnormal pulmonary venous return; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
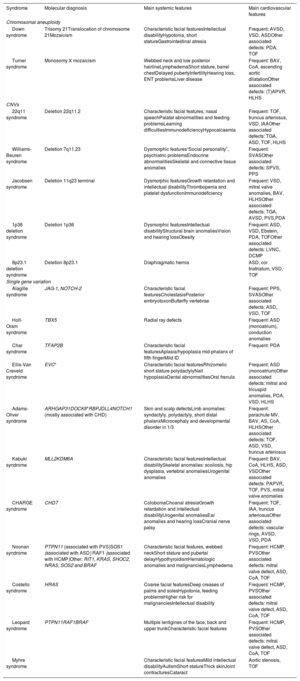
Overview of the main systemic and cardiovascular features of genetic syndromes associated with CHD
| Syndrome | Molecular diagnosis | Main systemic features | Main cardiovascular features |
|---|---|---|---|
| Chromosomal aneuploidy | |||
| Down syndrome | Trisomy 21Translocation of chromosome 21Mozaicism | Characteristic facial featuresIntellectual disabilityHypotonia, short statureGastrointestinal atresia | Frequent: AVSD, VSD, ASDOther associated defects: PDA, TOF |
| Turner syndrome | Monosomy X mozaicism | Webbed neck and low posterior hairlineLymphedemaShort stature, barrel chestDelayed pubertyInfertilityHearing loss, ENT problemsLiver disease | Frequent: BAV, CoA, ascending aortic dilatationOther associated defects: (T)APVR, HLHS |
| CNVs | |||
| 22q11 syndrome | Deletion 22q11.2 | Characteristic facial features, nasal speechPalatal abnormalities and feeding problemsLearning difficultiesImmunodeficiencyHypocalcaemia | Frequent: TOF, truncus arteriosus, VSD, IAAOther associated defects: TGA, ASD, TOF, HLHS |
| Williams-Beuren syndrome | Deletion 7q11.23 | Dysmorphic features“Social personality”, psychiatric problemsEndocrine abnormalitiesSkeletal and connective tissue anomalies | Frequent: SVASOther associated defects: SPVS, PPS |
| Jacobsen syndrome | Deletion 11q23 terminal | Dysmorphic featuresGrowth retardation and intellectual disabilityThrombopenia and platelet dysfunctionImmunodeficiency | Frequent: VSD, mitral valve anomalies, BAV, HLHSOther associated defects: TGA, AVSD, PVS,PDA |
| 1p36 deletion syndrome | Deletion 1p36 | Dysmorphic featuresIntellectual disabilityStructural brain anomaliesVision and hearing lossObesity | Frequent: ASD, VSD, Ebstein, PDA, TOFOther associated defects: LVNC, DCMP |
| 8p23.1 deletion syndrome | Deletion 8p23.1 | Diaphragmatic hernia | ASD, cor triatriatum, VSD, TOF |
| Single gene variation | |||
| Alagille syndrome | JAG-1, NOTCH-2 | Characteristic facial featuresCholestasisPosterior embryotoxonButterfly vertebrae | Frequent: PPS, SVASOther associated defects: ASD, VSD, TOF |
| Holt-Oram syndrome | TBX5 | Radial ray defects | Frequent: ASD (monoatrium), conduction anomalies |
| Char syndrome | TFAP2B | Characteristic facial featuresAplasia/hypoplasia mid-phalanx of fifth fingerMild ID | Frequent: PDA |
| Ellis-Van Creveld syndrome | EVC* | Characteristic facial featuresRhizomelic short stature polydactylyNail hypoplasiaDental abnormalitiesOral frenula | Frequent: ASD (monoatrium)Other associated defects: mitral and tricuspid anomalies, PDA, VSD, HLHS |
| Adams-Oliver syndrome | ARHGAP31DOCK6*RBPJDLL4NOTCH1 (mostly associated with CHD) | Skin and scalp defectsLimb anomalies: syndactyly, polydactyly, short distal phalanxMicrocephaly and developmental disorder in 1/3 | Frequent: parachute MV, BAV, AS, CoA, HLHSOther associated defects: TOF, ASD, VSD, truncus arteriosus |
| Kabuki syndrome | MLL2KDM6A | Characteristic facial featuresIntellectual disabilitySkeletal anomalies: scoliosis, hip dysplasia, vertebral anomaliesUrogenital anomalies | Frequent: BAV, CoA, HLHS, ASD, VSDOther associated defects: PAPVR, TOF, PVS, mitral valve anomalies |
| CHARGE syndrome | CHD7 | ColobomaChoanal atresiaGrowth retardation and intellectual disabilityUrogenital anomaliesEar anomalies and hearing lossCranial nerve palsy | Frequent: TOF, IAA, truncus arteriosusOther associated defects: vascular rings, AVSD, VSD, PDA |
| Noonan syndrome | PTPN11 (associated with PVS)SOS1 (associated with ASD) RAF1 (associated with HCMP)Other: RIT1, KRAS, SHOC2, NRAS, SOS2 and BRAF | Characteristic facial features, webbed neckShort stature and pubertal delayHypothyroidismHematologic anomalies and malignanciesLymphedema | Frequent: HCMP, PVSOther associated defects: mitral valve defect, ASD, CoA, TOF |
| Costello syndrome | HRAS | Coarse facial featuresDeep creases of palms and solesHypotonia, feeding problemsHigher risk for malignanciesIntellectual disability | Frequent: HCMP, PVSOther associated defects: mitral valve defect, ASD, CoA, TOF |
| Leopard syndrome | PTPN11RAF1BRAF | Multiple lentigines of the face, back and upper trunkCharacteristic facial features | Frequent: HCMP, PVSOther associated defects: mitral valve defect, ASD, CoA, TOF |
| Myhre syndrome | Characteristic facial featuresMild intellectual disabilityAutismShort statureThick skinJoint contracturesCataract | Aortic stenosis, TOF | |
ASD, atrial septal defect; AVSD, atrioventricular septal defect; BAV, bicuspid aortic valve; CHD, congenital heart defects; CoA, coarctation of the aorta; DCMP, dilated cardiomyopathy; HCMP, hypertrophic cardiomyopathy; HLHS, hypoplastic left heart syndrome; IAA, interrupted aortic arch; LVNC, left ventricular non compaction; PAPVR, partially abnormal pulmonary venous return; PDA, persistent ductus arteriosus; PVS, pulmonary valve stenosis; SVAS, supravalvular aortic stenosis; (T)APVR, (total) abnormal pulmonary venous return; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
Gene identification in isolated CHD has been hampered by several factors: first, defects in different genes may result in similar phenotypes, and different phenotypes may result from defects in the same gene. Second, especially in sporadic cases, the CHD may have a multifactorial cause. Despite high throughput molecular screening, unraveling multifactorial disease is still in its infancy. Recent advances in polygenic risk scores have been proposed for familial cancer and cardiomyopathies. It is to be expected that this will also hold true for CHD.
PRACTICAL APPLICATIONS OF GENETIC EVALUATION IN CHDAlong with increasing knowledge, the clinical impact of genetic testing in CHD shows continuous expansion. However, to incorporate genetic testing as part of the standard care of patients with CHD, several considerations deserve attention.
First, genetic testing should definitely be considered in specific subgroups of patients: those with syndromic features and those with multiple affected family members are most likely to be affected by an underlying genetic problem. Second, genetic testing and counseling should be tailored to the individual patient. The selection of the genetic test, as well as the appropriate timing, should be based on an individual basis. Third, when a genetic cause of CHD is identified, this should be accompanied by counseling to discuss appropriate management of the patient and his/her family and—when indicated—the recurrence risk.
Last, but definitely not least, genetic testing needs to take psychological, social and cultural aspects into account. The decision to proceed with such testing should be a well-informed decision in which the patient has the final word.
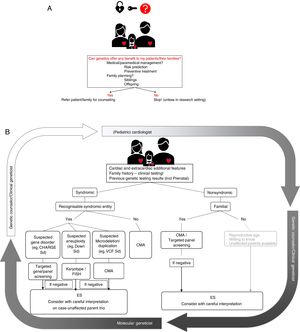
Some of these issues are discussed in more detail below and are illustrated in figure 1.
 cardiologists in collaboration with clinical geneticists and genetic counselors will check for additional cardiac and clinical features to rule out syndromic entities. Based on this, subsequent appropriate genetic testing will be set up in a stepwise approach. Exome sequencing is a final step that needs careful consideration and interpretation. Results will be relayed to the clinician and eventually to patients with appropriate counseling. CMA, chromosomal microarray analysis; ES, exome sequencing; FISH, fluorescent in situ hybridization; Sd, syndrome; VCF, velocardiofacial syndrome.")
The process of genetic evaluation in congenital heart disease A: carefully weigh the possible benefit of genetic assessment. B: algorithm for clinical/molecular testing. In a first step, (pediatric) cardiologists in collaboration with clinical geneticists and genetic counselors will check for additional cardiac and clinical features to rule out syndromic entities. Based on this, subsequent appropriate genetic testing will be set up in a stepwise approach. Exome sequencing is a final step that needs careful consideration and interpretation. Results will be relayed to the clinician and eventually to patients with appropriate counseling. CMA, chromosomal microarray analysis; ES, exome sequencing; FISH, fluorescent in situ hybridization; Sd, syndrome; VCF, velocardiofacial syndrome.
A first situation in which genetic testing should be considered, is in case of other extracardiac abnormalities, suggesting a syndromic entity. To correctly identify children and adults in this group, an accurate review of the medical history and extensive phenotypic characterization by a clinical geneticist is essential. Moreover, phenotypical screening of first degree family members can be necessary to identify syndromic features. Clinical manifestations that should trigger suspicion of an underlying syndromic problem are intellectual disability or sensory deficits, the presence of dysmorphic features and/or small stature, and the association of other congenital or endocrine disorders.15,60
A second situation in which genetic evaluation should be considered is when multiple family members are affected. Familial forms of CHD represent a small number of all CHDs, as reflected in a Danish population study where only 2.2% of the CHD were familial.4 Nevertheless, high diagnostic yields can be achieved in some families as shown by cases of familial supravalvular aortic stenosis in which genetic involvement can be found in 85% of families.61
Genetic testing may also be considered in neonates and infants with CHD. Genetic factors are important determinants of neurodevelopment and extracardiac lesions in children with CHD.62,63 Knowledge of this genetic predisposition might improve the long-term outcome of these children.
In addition to the indications mentioned above, adult patients with CHD and an active desire to have children (both men and women) are best referred for genetic counseling and possible testing. The advent of new preconceptional and prenatal techniques allows the preclusion of transmission of the CHD to the next generation. If there is an unknown underlying genetic disorder, genetic counseling is still very important to estimate the recurrence risk.
As already mentioned, adult patients with CHD who underwent genetic evaluation with older techniques might benefit from a reevaluation and retesting.
Tailoring genetic testingSelecting the most appropriate genetic test for each individual CHD patient is very important and requires close collaboration between clinical and molecular geneticists. It has been shown that a careful pretest review by a genetic counselor in consultation may reduce the proportion of inappropriate tests by 26%.64 The choice of technique during genetic diagnosis is highly dependent on the clinical presentation and family history. Figure 1 shows a flowchart on how to tailor these techniques to a specific patient.
Actionability of genetic findingsWhen referring a patient for genetic counseling and testing, the key question is always whether identification of a genetic defect can be of benefit to the patient or his/her family. In this respect, the following arguments can be taken into account:
Improved managementKnowledge of an underlying genetic problem can be important to diagnose and improve management of extracardiac complications in children and adults with CHD. For example, patients with 22q11 deletion may have decreased T cell immunity and therefore be at risk of severe infectious diseases; patients with Alagille syndrome can suffer from ophthalmologic and liver complications; children with Noonan syndrome have short stature and may benefit from growth hormone therapy.
Some genetic defects have been associated with an increased risk of other cardiovascular complications. A known example is the association between ASD and conduction disorders in those patients carrying a pathogenic variant in NKX2.5. These patients are more likely to develop atrioventricular block, ventricular dysfunction, and sudden cardiac death.65 Various types of CHD have been associated with genes causing familial cardiomyopathy. Some examples are the ACTC1, MYH6, and MYBPC3 genes.66–68
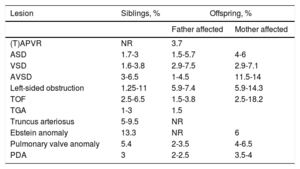
Recurrence risk and implications for other family membersWhen discussing recurrence risk, knowledge of an underlying genetic entity is essential. Risk estimates will also vary according to the setting of siblings or offspring and, in some cases, risks differ for fathers and mothers. If there is a known genetic disorder, recurrence risk in a sibling will greatly depend on the type of inheritance and on the de novo character of the anomaly. For those genetic disorders with an autosomal recessive pattern, the recurrence risk is 25%. For those with an autosomal dominant pattern, the recurrence risk will be 50% if one of the parents is affected and up to 1% in case of a de novo variation.69 For adult patients with CHD with a known genetic condition, the recurrence risk for the offspring will be 50% if the disorder is autosomal dominant. For those patients with autosomal recessive anomalies, the recurrence risk for children is similar to that in the general population, but each child will be carrier of 1 allele with the anomaly. If no underlying genetic anomaly is found, the recurrence risk is still higher than in the general population. Considering all CHD together, the recurrence risk for siblings is estimated at 2.1% and for the offspring at 4.4%, with women in general having a higher recurrence rate than men.70Table 4 summarizes the known epidemiological recurrence risk for the most common CHD in the absence of a known underlying genetic cause. Some lesions such as heterotaxia, right ventricular outflow track obstruction and atrioventricular septal defect, present higher familial clustering. The recurrence risk is 80-fold to 25-fold higher than in the general population.4
Recurrence risk of CHD in siblings and offspring of patients with nonsyndromic CHD without a known molecular defect5,70–74
| Lesion | Siblings, % | Offspring, % | |
|---|---|---|---|
| Father affected | Mother affected | ||
| (T)APVR | NR | 3.7 | |
| ASD | 1.7-3 | 1.5-5.7 | 4-6 |
| VSD | 1.6-3.8 | 2.9-7.5 | 2.9-7.1 |
| AVSD | 3-6.5 | 1-4.5 | 11.5-14 |
| Left-sided obstruction | 1.25-11 | 5.9-7.4 | 5.9-14.3 |
| TOF | 2.5-6.5 | 1.5-3.8 | 2.5-18.2 |
| TGA | 1-3 | 1.5 | |
| Truncus arteriosus | 5-9.5 | NR | |
| Ebstein anomaly | 13.3 | NR | 6 |
| Pulmonary valve anomaly | 5.4 | 2-3.5 | 4-6.5 |
| PDA | 3 | 2-2.5 | 3.5-4 |
ASD, atrial septal defect; AVSD, atrioventricular septal defect; CHD, congenital heart defects; NR, not reported; PDA, persistent ductus arteriosus; (T)APVR, (total) abnormal pulmonary venous return; TGA, transposition of the great arteries; TOF, tetralogy of Fallot; VSD, ventricular septal defect.
Another important aspect in terms of counseling family members is the need for further clinical assessment. For some lesions, such as LVOT obstruction, there is known variation in the clinical spectrum ranging from an asymptomatic BAV to a severe hypoplastic left heart syndrome.75 In one study, the relative risk of having a BAV in a parent or a sibling of a patient with a LVOT obstructive lesion was 5.05 (95% confidence interval, 2.2-11.7).11 This high incidence together with the fact that many of the complications of left-sided lesions are treatable or preventable, led to the rationale that first degree family members of patients with LVOT obstruction should undergo echocardiographic assessment.76 Currently no systematic screening of family members is recommended for other forms of nonsyndromic CHD.
Prenatal diagnosis and fetal screeningPrenatal diagnosis is possible whenever a genetic cause of the CHD has been identified. In this case, transmission to the next generation can be prevented through preimplantation diagnosis, a technique based on in vitro fertilization, which occurs before pregnancy and selects the unaffected embryos.
An alternative is prenatal testing, which conventionally implies chorionic villus or amniotic fluid sampling in the early stages of pregnancy. In more recent years, noninvasive prenatal testing (NIPT) has emerged as a noninvasive alternative for prenatal testing. NIPT was primarily developed to detect trisomy 21 in the fetus early in pregnancy with high specificity (> 99%) and sensitivity (> 99%) in the absence of fetal anomalies. During pregnancy, cells from the placenta (containing fetal DNA) lyse into the maternal circulation. The test is based on the relative number of reads that map to a certain chromosome in maternal plasma (cell free DNA). Hence, the test can detect other aneuploidies such as trisomy 13 and 18, Klinefelter (47,XXY), Turner (45,X0), or triple X (47,XXX) syndrome, albeit with a somewhat lower sensitivity and specificity. Technological fine tuning will eventually render NIPT suitable to detect de novo variants (both CNVs and SNVs)77,78 and targeted testing of inherited variants.79 It goes without saying that broad scale application of NIPT requires careful consideration of ethical issues. In both instances of prenatal testing, termination of pregnancy may be considered if the results of the test are abnormal test.
Some couples may choose not to undergo prenatal diagnosis, in which case, the possibility of genetic screening in the newborn should be discussed. If no underlying genetic cause of the CHD has been identified or no prenatal testing has been performed, fetal echocardiography is recommended. This should be performed in a specialized center at 18 to 20 weeks of pregnancy.80
CONCLUSIONGenetic evaluation of patients with CHD is being conducted on an increasingly large scale and will in many cases undoubtedly help us to optimize (para)medical management in individual patients and their families. Moreover, knowledge of genetics further helps us to understand the underlying pathophysiology of these conditions, which will certainly contribute in the long-term to developing more targeted treatments.
Genetic evaluation should be done correctly in each patient/family with careful counseling prior to testing as well as upon communication of any results. Implementation of a correct strategy has already clearly demonstrated that this leads to more efficient testing, greater patient satisfaction, and more correct medical management
FUNDINGJ. De Backer and B. Callewaert are funded as senior clinical researchers by the Flanders Research Foundation. J. De Backer is a recipient of the Grant for Medical Research from the Baillet Latour Fund.
CONFLICTS OF INTERESTThe authors have no conflicts of interest.