
Hereditary transthyretin amyloidosis (ATTRv) is a multisystemic disease with autosomal dominant inheritance and variable penetrance, caused by mutations in the TTR gene. More than 120 genetic variants associated with the disease have been described, some of them considered endemic to certain geographic areas and with a strong genotype-phenotype correlation.1,2 The heart and nervous system are the organs most often affected in ATTRv. The cardiac manifestations are usually heart failure, conduction disturbances, or atrial fibrillation, whereas the neurological features include symmetrical and distal sensory-motor polyneuropathy, often associated with dysautonomia symptoms.1,2 Until recently, the only therapeutic option for ATTRv was liver transplant; however, specific drugs proven to increase survival are now available for these patients.3
The p.Ser43Asn variant of the TTR gene is a very rare cause of ATTRv, reported in only 7 cases to date.4–6 Because it is so uncommon, the available information does not suffice to predict the expected clinical course of the disease or the type of follow-up management to use in carriers of the variant.
After identification of 3 independent families affected by ATTRv caused by the p.Ser43Asn variant, we endeavored to study the characteristics and clinical course of this condition, and determine possible geographic areas where the variant could be endemic.
A retrospective observational study was carried out in all carriers of the p.Ser43Asn variant followed up in our center. The study was approved by the Puerta de Hierro Hospital Ethics Committee, which did not require informed consent from the participants. The variant was detected by sequencing the 4 exons of the TTR gene in index cases and the affected exon in relatives. Cardiac involvement was established based on left ventricular thickness ≥12mm or Perugini grade 2 or 3 uptake of 99mTc-DPD on cardiac scintigraphy, after exclusion of blood cell dyscrasia.3 Neurological involvement was determined by the presence of neurological symptoms consistent with the disease, together with abnormal neurological test findings.1 The variables collected included the patients’ clinical characteristics and results of complementary tests in the first evaluation, and the clinical course and events that occurred during follow-up. Quantitative variables are presented as the median [interquartile range] and categorical variables as the number and percentage.
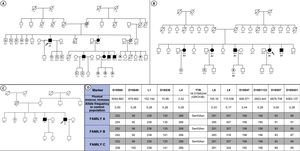
Eight individuals from 3 independent families (3 probands and 5 relatives: 4 men; age, 29-60 years) were identified as carriers of the p.Ser43Asn variant (figure 1A-C). The characteristics of these individuals are shown in table 1. The 3 families were not related, but were all originally from southern Ecuador, and more specifically from the Loja region. In the first evaluation, all probands were found to have both cardiac and neurological involvement, whereas among the relatives, only 1 showed a mixed phenotype, 2 had neuropathy alone, 1 cardiomyopathy alone, and 1 was an asymptomatic carrier. In addition, 4 patients reported a history of carpal tunnel syndrome in the first evaluation. Median age at disease onset was 49.8 [44.0-58.8] years. The heart condition in all probands was characterized by advanced symptoms (New York Heart Association II-III). None of the patients had atrial fibrillation or pacemaker implantation before the first evaluation. All those with neurological involvement showed isolated sensory symptoms (Polyneuropathy Disability score [PND] I), with a median Neuropathy Impairment Score (NIS) of 3 [2-11].
 and haplotype study (D). Squares indicate men and circles women. White symbols represent healthy individuals, black symbols individuals with a diagnosis of ATTRv, and gray symbols individuals with suspected ATTRv. White symbols with a central dot are asymptomatic carriers. White symbols with an N represent individuals testing negative for Ser43Asn. Individuals A1, B1 and C1 are the probands in each family. Diagonal lines indicate those who have died. ATTRv, hereditary transthyretin amyloidosis.")
Genealogic trees of the families included (A-C) and haplotype study (D). Squares indicate men and circles women. White symbols represent healthy individuals, black symbols individuals with a diagnosis of ATTRv, and gray symbols individuals with suspected ATTRv. White symbols with a central dot are asymptomatic carriers. White symbols with an N represent individuals testing negative for Ser43Asn. Individuals A1, B1 and C1 are the probands in each family. Diagonal lines indicate those who have died. ATTRv, hereditary transthyretin amyloidosis.
Characteristics of patients with ATTRv due to the Ser43Asn variant included in the study
| Patient | A1 | B1 | B2 | B3 | B4 | B5 | B6 | C1 |
|---|---|---|---|---|---|---|---|---|
| Sex | Man | Woman | Woman | Woman | Man | Man | Woman | Man |
| Age at diagnosis, y | 50 | 47 | 60 | 58 | 51 | 47 | 29 | 41 |
| Country of origin | Ecuador | Ecuador | Ecuador | Ecuador | Ecuador | Ecuador | Ecuador | Ecuador |
| Age at onset of symptoms, y | 50 | 47 | 61 | 59 | 52 | — | — | 40 |
| Cardiologic involvement | Yes 99mTc-DPD (+) | Yes 99mTc-DPD (+) | No 99mTc-DPD (–) | No 99mTc-DPD (–) | Yes 99mTc-DPD (+) | Yes 99mTc-DPD (+) | No | Yes 99mTc-DPD (+) |
| Functional class | NYHA II | NYHA II | — | — | NYHA I | NYHA I | — | NYHA III |
| Neurologic involvement | PND I, NIS 11 | PND I, NIS 2 | PND I, NIS 4 | PND I, NIS 11 | PND I, NIS 0 | No | No | PND I, NIS 2 |
| Other manifestations | Unilateral CTS | — | Bilateral CTS | — | Bilateral CTS | — | — | Unilateral CTS |
| Treatment | Tafamidis | Tafamidis | Tafamidis | Tafamidis | Tafamidis | — | — | Patisiran |
| Follow-up, mo | 36.1 | 17.0 | 15.6 | 8.1 | 18.9 | 3.3 | 4.4 | 10.8 |
| Events | HF admissions, de novo AF, complete AVB | HF admissions, de novo AF, Shock/HF | — | — | HF admission | — | — | HF admission, complete AVB |
AVB, atrioventricular block; AF, atrial fibrillation; HF, heart failure; NIS, Neuropathy Impairment Score; NYHA, New York Heart Association; PND, Polyneuropathy Disability score; CTS, carpel tunnel syndrome; 99mTc-DPD, scintigraphy with 99m technetium and 3,3-diphosphono-1,2-propanedicarboxylic acid
For first-line treatment, 5 patients received tafamidis and 1 started patisiran, as the diagnosis was made in an advanced phase of the disease. After a median follow-up of 13.2 [6.2-17.9] months, 4 patients required hospital admission for heart failure, 2 developed de novo atrial fibrillation, and 2 required pacemaker implantation due to complete atrioventricular block. One of the patients progressed to refractory cardiogenic shock and required biventricular mechanical circulatory support as bridging therapy to heart transplant. The patient had a poor clinical response following transplant and died due to infectious complications during the postoperative period. The PND score did not worsen in any patient during follow-up. A haplotype study performed with DNA samples from the 3 families showed concordance in 11 genetic markers around the p.Ser43Asn variant, indicating that the families likely arose from a recent common ancestor (12.4 generations; 95% confidence interval [95%CI], 5.4-28.9) (figure 1D).
Despite the limitations of an observational study with a small patient sample and short follow-up, the findings obtained here indicate that ATTRv due to the p.Ser43Asn variant is associated with a mixed cardiologic and neurologic phenotype with onset after the age of 40 years. These results contrast with those of the previously reported cases4–6 in which neurological involvement was less prevalent, and highlight the importance of conducting an in-depth neurological evaluation to enable prompt initiation of disease-modifying treatment. As was seen in the previous descriptions, the cardiologic condition determines the prognosis, with aggressive progression to advanced heart failure, whereas the neurological condition is mild. In light of the aggressive clinical course and documented age of onset, close follow-up is recommended for carriers of the variant, starting at age 30 to 35 years.
From the epidemiological viewpoint, the variant was identified in 3 independent families from the province of Loja, Ecuador, with a concordant haplotype study, which suggests an endemic focus associated with a founder effect. Some of the reported cases also come from Latin America (Ecuador and Peru),4,5 in line with our findings. We believe that detection of the cases described here outside the country of origin may be related to limited access to a clinical diagnosis and genetic testing for ATTRv in Ecuador. Although additional studies are needed to determine the prevalence of ATTRv due to p.Ser43Asn in Ecuador, the findings reported here should raise suspicion of this condition in patients from the Loja region or from Ecuador showing signs and symptoms consistent with ATTRv.
FUNDINGThis study was funded by the Carlos III Health Institute (ISCIII) through the projects PI18/0765 and PI20/01379 (co-financed by the European Regional Development Fund/European Social Fund, “A way of making Europe/The ESF invests in your future”). The National Center for Cardiovascular Research (CNIC) is dependent on the Carlos III Health Institute, the Ministry of Science and Innovation (MCIN), and the Pro-CNIC Foundation, and is accredited as a Severo Ochoa Center of Excellence (CEX2020-001041-S).
AUTHORS’ CONTRIBUTIONSE. Porres-López and F. de Frutos are the first authors.
E. Porres-López and F. de Frutos collected and analyzed the data, and wrote the first draft. L. Silva-Hernandez, L. Galán, and E. González-Lopez collected the data, reviewed the work undertaken, and made relevant intellectual contributions. Pablo Garcia-Pavía supervised the study, obtained funding, and participated in writing the manuscript.
CONFLICTS OF INTERESTP. García-Pavía is an associate editor of the Spanish Journal of Cardiology. The editorial procedure established by the Journal has been followed to guarantee impartial management of the manuscript.
We thank Elvira Ramil from the Molecular Biology and DNA Sequencing Unit of the Puerta de Hierro-Segovia de Arana Research Institute (Madrid, Spain), as well as Juan Pablo Ochoa and Enrique Lara Pezzi from the CNIC (Madrid, Spain) for their collaboration in the haplotype study.