Keywords
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is an inherited heart disease characterized by thickening of the myocardial wall in the absence of other causes of left ventricular hypertrophy.1 It is one of the most common causes of sudden death in young persons and the leading cause of inherited cardiovascular disease, with an estimated prevalence of one case for each 500 persons.2
To date, over 400 mutations causing HCM have been identified in 11 genes that encode for cardiac sarcomere proteins.3 One of these genes is MYBPC3, which encodes for myosin-binding protein C. Mutations in the MYBPC3 gene have traditionally been considered to cause late-onset HCM, with less ventricular hypertrophy and a better prognosis than HCM caused by mutations in the other sarcomere genes.4-6
We report the case of a family with several members having HCM and in which there was a high incidence of sudden death. Various members of the family had a mutation in the MYBPC3 gene that has not been described previously.7
METHODS
We studied the living members in a branch of a family which had previously: multiple family history of sudden death and heart disease (Figure 1). The study included a physical examination, electrocardiogram and echocardiogram. Those members with HCM also underwent an ergometry test and 48-hour electrocardiogram.

Figure 1. Family genealogical tree. Circle indicates female. Square, male. Red symbols, members of the affected family with heart disease or who had died suddenly. Brown symbols, members without heart disease. Symbols with a circle inside, members of the family who were carriers of the mutation without heart disease at the time of evaluation. Symbols with a diagonal line, deceased members. Arrow, index patient.
After establishing the diagnosis of HCM in the index patient, the elder of her daughters, who was 34 years old, was studied in Norway, where she lived, and also diagnosed with non-obstructive HCM. After obtaining written, informed consent, we undertook a genetic study in this patient by exon sequencing of the ß myosin heavy-chain gene (MYH7), myosin-binding protein C (MYBPC3), troponin I (TNNI3), troponin T (TNNT2), myosin regulatory light chain 2 (MYL2), and the myosin essential light chain 1 (MYL3). The genes studied are responsible for almost all known cases of HCM involving a mutation.8
After identifying a mutation in the MYBPC3 gene, we undertook a genetic study in the index patient and in those first-degree members of the families with persons having HCM or who had died suddenly (Figure 1; subjects II-1, III-1, III-2, IV-2, IV-3, IV-4, IV-7, IV-8, and IV-9). This genetic study consisted of polymerase chain reaction (PCR) amplification of target DNA from exon 2 and the adjacent intron regions of the MYBPC3 gene.
The primers and methods used are available on request to the address for correspondence given above. The genetic study was approved by the Ethics Committee of La Paz Hospital.
RESULTS
Clinical Study
Index Patient
The index patient was a 53-year old woman with a long history of hypertension and smoking who was referred to our center for progressive dyspnea on effort. The initial examination showed radiologic signs of cardiomegaly and electrocardiographic signs of left ventricular hypertrophy. An echocardiogram showed severe hypertrophy of the anterior, anteroseptal and septal walls with an interventricular septal thickness of 25 mm, normal contractility, E wave velocity of 0.71 ms, E wave deceleration time of 130 ms and absence of a subaortic gradient, findings compatible with the diagnosis of non-obstructive HCM with advanced diastolic dysfunction. After documenting nonsustained ventricular tachycardia (NSVT) on two 48 h electrocardiographic recordings and given the family history of sudden death, the patient underwent surgery for the implantation of an implantable automatic defibrillator (IAD).
Family History
The paternal grandfather of the index patient, her father and one of her paternal uncles had died suddenly around the age of 50 years (52, 49, and 48 years of age, respectively). Two brothers had been diagnosed with heart disease. The first died suddenly aged 32 years with no children and the second, who was the only one with an established diagnosis of HCM, died aged 42 years after having a cerebral hemorrhage whilst on the waiting list for a heart transplant. The daughter of the index patient (mentioned earlier), diagnosed with HCM at the age of 34 years, showed NSVT on the ambulatory electrocardiographic recording, and accordingly also underwent surgery for an IAD. The other members of the family who were studied remained asymptomatic and had normal echocardiograms (Figure 1).
Genetic Study
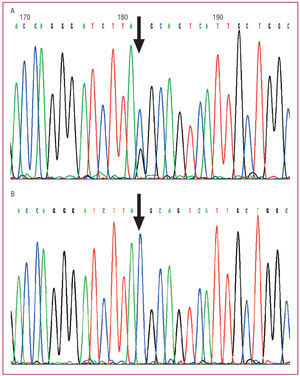
The index patient and her daughter who had HCM were found to be heterozygous for the Y79X mutation in exon 2 of MYBPC3. The substitution of cytosine by guanine at nucleotide 269 of one of the alleles of MYBPC3 (Figure 2) (TAC > TAG) gives rise to a stop in codon 79, such that instead of encoding for the amino acid tyrosine (TAC) it is transformed into a stop codon (TAG). This mutation invariably causes the interruption of the translation of mRNA at this point, resulting in the synthesis of an incomplete protein.

Figure 2. Sequencing of exon 2 of MYBPC3 in A: index patient (arrow in Figure 1) and B: non-carrier daughter of the index patient (* in Figure 1). A: two peaks are shown, representing both cytosine (blue) and guanine (black) at the point corresponding to nucleotide 269 of MYBPC3. This double peak indicates that in one of the alleles, the TAC codon, which encodes for tyrosine 79 of the protein, is transformed to TAG (stop or nonsense codon). B: the same point in MYBPC3 shows a single peak (cytosine, blue), which corresponds to 2 normal alleles for the TAC codon of tyrosine 79.
Another three members of the family with no clinical signs of heart disease (subjects IV-3, IV-4 and IV-8, aged 19, 15, and 28 years, respectively) had the same mutation in exon 2 MYBPC3. Subjects II-1, III-2, IV-2, IV-7, and IV-9 did not have this mutation (Figure 1).
DISCUSSION
Since the initial identification in 1990 of one of the genes related with HCM,9 the progressive knowledge of the genetic basis of the disease has completely changed the care of patients with HCM and their families.
The genetic study undertaken in the family presented here led to the identification of a new mutation in MYBPC3. This mutation is associated with an unfavorable prognosis, with development of the disease and sudden death between the fourth and sixth decades of life.
Despite the supposedly benign course of secondary HCM with mutations in MYBPC3,4-6 the presentation in this family supports the findings of other studies, which have related the course of the disease to the type of mutation in this gene.10 Thus, patients with stop mutations in MYBPC3 (such as that described in this paper) present the disease at an earlier age and require more aggressive treatment (septal ablation and implantation of a defibrillator) than patients with mutations that just give rise to changes in the amino acid sequence.10 An IAD was implanted in the index patient of this family and in her daughter, who had HCM.
Although the mutation reported is likely to be the cause of the HCM in this family, it cannot be stated categorically, as this mutation has not been found in other families with HCM, and not all the genes responsible for HCM were studied. The influence of alterations in other genes could play a fundamental role in the clinical manifestations of the disease and could, in part, explain the differences seen in the clinical course between the various members of the family in question. It was notable that all the members of the family who had died were male and that they had died at an earlier age than the index patient when she was diagnosed.
The identification of the causative mutation in the asymptomatic relatives enabled us to clearly determine those persons who were susceptible to the future development of HCM and those who did not need a specific follow-up, as they were not carriers of the mutation.11 In this family, the sister of the index patient, one daughter, a nephews and a niece, and the children of all these do not require follow-up in the future (Figure 1).
Identification of a mutation in patients with HCM or asymptomatic carriers of a mutation can enable early genetic counseling,12 and if the mutation has been described previously, it enables the likely course of the disease to be elucidated. All the carriers of the mutation in this family were offered genetic counseling in order to plan their children. Future patients with HCM associated with this same mutation in MYBPC3 could be informed about the possible course of the disease based on the analysis of the family described here, with the obvious limitations described above.
The process to follow in persons who have the mutation but who have not developed HCM remains to be established.13 A recent study highlighted the superiority of cardiac magnetic resonance imaging (MRI) over traditional echocardiography for the diagnosis of HCM in healthy carriers of the mutation.14 The three carriers of the mutation who did not have heart disease in this family (aged 19, 15, and 28 years) had no signs that were compatible with HCM in the cardiac MRI study performed. We plan to follow up these patients with ECG, echocardiograms and cardiac MRI yearly as long as they remain asymptomatic.
In summary, this study describes a previously unknown mutation associated with HCM of poor prognosis, and shows the clinical usefulness of undertaking a genetic study in patients with HCM.
This study was financed in part by a MAPFRE Medicina 2005 grant.
Correspondence: Dr. P. García-Pavía.
Servicio de Cardiología. Hospital Universitario Puerta de Hierro.
San Martín de Porres, 4. 28035 Madrid. España.
E-mail: pablogpavia@yahoo.es
Received March 27, 2006.
Accepted for publication August 30, 2006.