Keywords
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is an autosomal dominant hereditary disease caused by over 500 mutations in 11 different genes.1-4 Information about the relationship between genotype and phenotype and the prognosis associated with various mutations in the troponin T gene (TNNT2) is limited and at times contradictory.4,5-13 The aim of this study was to identify mutations in the TNNT2 gene and study the correlations between the genotype and the phenotype.
METHODS
Blood samples were studied from 127 consecutive patients with HCM (mean age, 50 [16] years; 66% men; maximum wall thickness, 18 [5] mm; 28% with obstruction). The patients came from 3 centers: the Hospital Universitario in A Coruña (60 patients), the Hospital Virgen de la Arrixaca (37 patients), and the Hospital General in Alicante (30 patients). Cardiac evaluation included ECG, echocardiogram, Holter, and an exercise stress test. All the patients gave written informed consent.
Each index patient provided 4 mL of blood, drawn into an EDTA tube. The DNA was extracted and amplified, and TNNT2 exons were analysed by screening with denaturing high-performance liquid chromatography (DHPLC). In the event of an anomalous profile, the exon was sequenced (sequencer: ABI310/ABI3130). Specific primers were designed for the amplification of the fragments of interest.
RESULTS
Eight different alterations were found in 107 exons. Three variants were considered to be causal mutations: Phe87Leu (not previously reported), Arg278Cys and Asp271Ile (Table 1). The Asp271Ile mutation was identified in a Galician family and the Arg278Cys mutation in one family from Murcia and another from Alicante. Both these mutations had already been associated with HCM. The Phe87Leu mutation was considered to be causative as it segregated with the disease, affected a highly conserved residue and was not identified in 140 healthy controls. Table 2 summarizes the clinical characteristics of the carriers.

Clinical Characteristics and Genotype-Phenotype Correlation
Phe87Leu
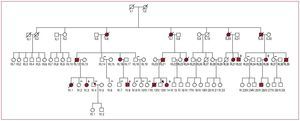
This mutation was identified in a family with history of sudden death (SD) (Figure 1). In total, 18 persons were affected, with 11 related deaths: 7 SD, 3 due to heart failure and 1 from stroke. Four of those who died were aged between 14 and 16 years and were asymptomatic ([IV.1], [IV.12], [III.31], and [III.39]). Of the 10 patients for whom an echocardiogram was available, 7 had mild hypertrophy (<16 mm); in 2 of these the echocardiogram did not show the hypertrophy and the diagnosis was made at autopsy. Two of the patients with mild hypertrophy had a restrictive pattern. Three patients had a wall thickness >20 mm (maximum, 27 mm [IV.8]). Only the index case [IV.14] had a gradient. Systolic function was conserved, except in one case (ejection fraction, 40% [III.21]). One of the affected members had recurrent syncope [IV.3]. In general, the younger affected members of the family were asymptomatic and over half the affected members aged between 40 and 50 years had dyspnea (NYHA II-III). The family and genetic study led to implantation of automated defibrillators for primary prevention in 2 affected members [III:36 and IV:3]. The mutation was not identified in 6 members with a normal phenotype.

Figure 1. Family tree of A8 with Phe87Leu mutation in TNNT2.
Arg278Cys
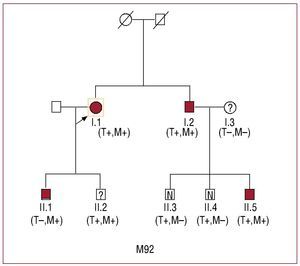
This mutation was identified in two families, M92 and A16 (Figure 2 and Table 2). Of the 8 carriers, 4 fulfilled the diagnostic criteria for HCM (A16: II:1; M92: I:1, I:2, II:5), 1 had a doubtful phenotype (M92: II:2) and 3 had a normal echocardiogram and ECG (M92: II:3 and II:4, A16: III:1) (Table 2). These families had no history of adverse events related with the disease.

Fig. 2. Family tree of M92 with Arg278Cys mutation in TNNT2. M indicates MYH7; T, TNNT2

One of the carriers in family M92 had very marked hypertrophy (40 mm) (II:5), which contrasted with the normal phenotype of this patient's siblings who were carriers. His father (I:2) had moderate hypertrophy and his mother (I:3) had mild hypertrophy. In order to attempt to clarify the relationship between genotype and phenotype in family M92, sequencing was made of another 8 sarcomeric genes (MYH7, MYBPC3, alpha tropomyosin, actin, TNNI2, TNNC1, and the myosin light chains). The patient with severe hypertrophy (II:5) carried an Asp 928Asn mutation in the beta myosin heavy chain (MYH7), which was also present in his father (I:2), aunt (I:1) and cousins (II:1) and (II:2). Three of the 4 carriers of the 2 mutations had a moderate or severe phenotype (2 adults and 1 young person). The mother of the case with severe hypertrophy (I:3) was negative for both mutations.
Asp271Ile
This mutation was identified in a family with no history of SD. The index case was diagnosed at the age of 44 years because of atypical pain and paroxysmal atrial fibrillation (II:2). His son had mild hypertrophy and an abnormal electrocardiogram at the age of 33 years (III:1). His daughter had an ostium secundum type atrial septal defect with no ventricular hypertrophy (III:2).
DISCUSSION
Hypertrophic cardiomyopathy is a heterogeneous disease in both its clinical presentation and its molecular basis. Only a few families have been reported with each of these mutations, and it is consequently difficult to draw conclusions about the relationship between genotype and phenotype that could enable their risk profile to be established.
We know that certain mutations are associated with a worse prognosis.4 The TNNT2 gene harbors some of the so-called malignant mutations. The prevalence of mutations in the TNNT2 gene in this sample of the Spanish population was 3%, similar to that found by other authors.3,6,7
In this report we describe for the first time the Phe87Leu mutation, identified in a family from Alicante. This mutation affects the binding site of troponin with alpha tropomyosin. The phenotype is characterized by mild hypertrophy, on occasions absent, and a very poor prognosis. A few cases posed serious diagnostic problems, which were able to be resolved from the genetic information.
The Arg278Cys mutation was identified in 2 families. This mutation has been reported in 11 families from various countries.4,6,7,10 The mutation affects an amino acid located at the binding site of tropomyosin and troponins I and C. Including our two families, there are now 36 carriers. The penetrance is incomplete, and hypertrophy, which becomes evident at advanced ages (50 years), is usually mild (17 mm). Reports exist of 11 SD, most in persons older than 60 years of age, although 2 cases occurred in young persons without ventricular hypertrophy.4 Six of the 11 SD happened in the same family, so there might have been some additional unidentified risk factor.6
We identified a family with the Asp271Ile mutation; only one case has been previously reported, and we do not have the clinical data.3 Three carriers in our family had a mild phenotype, and in one of these there was atrial communication. The data currently available do not allow the risk profile to be established.
Our study confirms that carriers of the mutations in TNNT2 can develop mild phenotypes, as has been indicated previously,4,5 and that mutations exist that are associated with a very high risk of SD, as is the case of the Phe87Leu mutation. In these cases, genetic analysis is a very useful clinical tool.
A few patients with severe phenotypes might carry double mutations, or the phenotype could be the consequence of the involvement of unknown modulating agents.3,13
We have also confirmed that some mutations in TNNT2, such as Arg278Cys, can cause late onset HCM. In these cases, the risk of SD may be greater in patients of a certain age.
In conclusion, mutations in the TNNT2 gene caused 3% of cases of HCM in our population. The Phe87Leu mutation is associated with mild hypertrophy and a high risk of SD. The Arg278Cys mutation is associated with mild hypertrophy, incomplete penetrance and a high risk in older carriers. The presence of double mutations should be suspected in persons with severe forms of the disease.
ACKNOWLEDGMENTS
This study was undertaken with the collaboration of doctors Gonzalo de la Morena, Francisco Ruiz, Eduardo Payá, María López, and Francisco Sogorb.
This study was undertaken with a grant from the Sociedad Española de Cardiología for clinical and basic research in cardiology, 2004. The study was partly funded by the Red de Investigación Cardiovascular RECAVA of the Instituto de Salud Carlos III (C03/01, RD06/0014/0017, RD06/0014/0018).
Correspondence: Dr. J.R. Gimeno.
Servicio de Cardiología. Hospital Universitario Virgen de la Arrixaca. Ctra. Murcia-Cartagena s/n. 30120 El Palmar. Murcia. España.
E-mail: jgimeno@secardiologia.es
Received October 21, 2008.
Accepted for publication February 18, 2009.