We report a family with many members affected by a form of incomplete Marfan syndrome (MS) called the MASS phenotype. The MASS phenotype consists of mitral valve prolapse (M), nonprogressive aortic root dilatation (A), musculoskeletal findings (S), and skin striae (S), thus resembling features of MS but not meeting the diagnostic criteria for this disease.1 Familial genetic analysis confirmed the pathogenicity of a nonsynonymous mutation (NP_000129.3:p.Pro1424Ser, NM_000138.4:c.4270C>T) in the fibrillin-1 gene (FBN1).
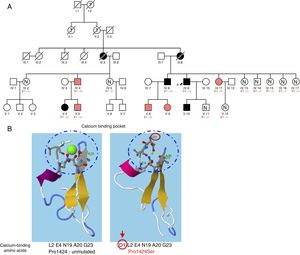
The index patients were 2 brothers who underwent aortic replacement for aortic root dilatation (patients IV.8 and IV. 9 in Figure 1A). These patients had a family history of sudden cardiac death, with postmortem examinations of their mother and aunt having revealed aortic dissection. The patients were assessed for ocular and locomotor function and underwent a thoracoabdominal computed tomography examination, and 1 of the patients was screened by high throughput sequencing targeting a 30-gene panel. A single study with this panel permitted cost-effective analysis of point mutations and copy number variants (CNV; large deletions and duplications) and elimination of other candidate syndromes.

A, Family tree. Patients represented by black shading meet the criteria for Marfan syndrome, whereas those represented by pink shading have the MASS phenotype. B, RaptorX in silico analysis of the calcium binding site in the EGF-like domain 24 of FBN1, comparing the unmutated form with the change caused by the Pro1424Ser mutation in FBN1. E1–/+, heterozygote for Pro1424Ser; E1–/–, noncarrier.
The genetic screen detected a missense mutation in a coding region in exon 34 of FBN1 (a single nucleotide change resulting in an amino acid alteration: p.Pro1424Ser). This mutation appears in the UMD-FBN1 mutation database in a single patient who met the criteria for MS; however, there was no recorded analysis of familial cosegregation, which is essential for confirming a mutation as pathogenic.1,2 No record of this mutation was found in other publicly available genotype databases, and searches of the ExAC and gnomAD population databases identified no mutation carriers, indicating a very low allele frequency in control populations. In silico studies with the RaptorX program showed that the affected proline residue (Pro1424) participates in hydrogen bonds required for the formation of the calcium binding site in this region (epidermal growth factor [EGF] like domain 24). The substitution of this proline by serine would alter the site and cause protein misfolding and disrupted function (Figure 1B).3
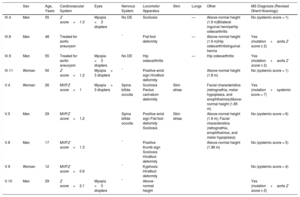
A complete familial analysis (Figure 1A) detected the c.4270C>T mutation in 9 family members manifesting variable signs of MS (Table 1). Aortic root dilatation was reported only in the probands and patient V.10, and the most frequent cardiovascular finding was mitral valve prolapse. Myopia> 3 diopters was recorded in 2 patients, and none of the patients had ectopia lentis. Almost all patients had skeletal alterations. Most had scoliosis, flat foot deformity, or above-normal height, whereas wrist or thumb signs were found in only 3 patients. No pathological findings were recorded in noncarrier relatives.
Phenotypic Characteristics of Family Members
| Sex | Age, Years | Cardiovascular System | Eyes | Nervous System | Locomotor Apparatus | Skin | Lungs | Other | MS Diagnosis (Revised Ghent Nosology) | |
|---|---|---|---|---|---|---|---|---|---|---|
| IV.4 | Man | 55 | Z score=1.3 | Myopia <3 diopters | No DE | Scoliosis | — | Above-normal height (1.9 m)Bilateral inguinal herniasHip osteoarthritis | No (systemic score = 1) | |
| IV.8 | Man | 48 | Treated for aortic aneurysm | * | Flat foot deformity | — | Above-normal height (1.9 m)Hip osteoarthritisInguinal hernia | Yes (mutation+aorta Z score ≥ 2) | ||
| IV.9 | Man | 55 | Treated for aortic aneurysm | Myopia <3 diopters | No DE | Hip osteoarthritis | — | Hip osteoarthritis | Yes (mutation+aorta Z score ≥ 2) | |
| IV.11 | Woman | 50 | Z score=1.2 | Myopia> 3 diopters | * | Positive wrist sign Hindfoot deformity | Above-normal height (1.8 m) | No (systemic score = 1) | ||
| V.4 | Woman | 26 | MVPZ score=1 | Myopia> 3 diopters | Spina bifida occulta | Scoliosis Pectus carinatum deformity | Skin striae | Facial characteristics (retrognathia, malar hypoplasia, and enophthalmos)Above-normal height (1.85 m) | Yes (mutation+systemic score = 7) | |
| V.5 | Man | 29 | MVPZ score=1.2 | Spina bifida occulta | Positive wrist sign Flat foot deformity Scoliosis | Skin striae | Above-normal height (1.9 m), Facial characteristics (retrognathia, enophthalmos, and malar hypoplasia) | No (systemic score = 6) | ||
| V.8 | Man | 17 | MVPZ score=1.3 | * | Positive thumb sign Scoliosis Hindfoot deformity | Above-normal height (1.96 m) | No (systemic score = 5) | |||
| V.9 | Woman | 12 | MVPZ score=0.9 | * | Kyphosis Hindfoot deformity | No (systemic score = 4) | ||||
| V.10 | Man | 29 | Z score=2.1 | Myopia <3 diopters | * | Above-normal height | Yes (mutation+aorta Z score ≥ 2) |
DE, dural ectasia; MVP, mitral valve prolapse.
Together, these data indicate familial cosegregation of the mutation with variable clinical manifestations. This behavior has been described previously for pathogenic mutations of this gene,4 and complete familial analyses should therefore be conducted at referral centers to obtain a better understanding of the phenotypic behavior of these gene variants.5 All the patients apart from V.4, IV.8, and IV.9 can be categorized as having the MASS phenotype because they do not meet one of the revised Ghent criteria for MS.1 Most of the carriers are young, which might explain the general absence of cardinal manifestations such as aortic root dilatation and ectopia lentis. Trans-acting mutations in FBN1 have been proposed to play a modifying role,4 and a recent study demonstrated that cis-regulatory variants can be present in the same allele as a disease-causing mutation, providing a possible explanation for the variable penetrance and expressivity observed in mutation carriers.6 Although c.4270C>T mutation carriers showed no signs of dural ectasia, spina bifida occulta was detected in 2 brothers (V.4 and V.5).
Another mutation affecting the same amino acid has been identified in several MS patients. However, the Pro1424Ala (g.48764814G>C) mutation has not been examined by cosegregation analysis, and this variant has been identified in 54 out of 277 166 individuals in gnomAD control populations, raising doubts about its pathogenicity.
In summary, the clinical and genetic study of this family confirms the association of the p.Pro1424Ser genetic variant in FBN1 with the development of the MASS phenotype and MS. Although the phenotypes generated by this genetic variant vary widely and are sometimes mild, it is important to monitor aortic diameter regularly in carriers of this mutation because it can be associated with aneurysm and dissection.
CONFLICTS OF INTERESTJ.P. Trujillo-Quintero and L. Montserrat are members of the genetic diagnostics company Health in Code.