Sustained monomorphic ventricular tachycardia (SMVT) in the setting of an anterior acute myocardial infarction (AMI) is rare. We present a case that illustrates the diagnostic, prognostic and therapeutic implications of this entity.
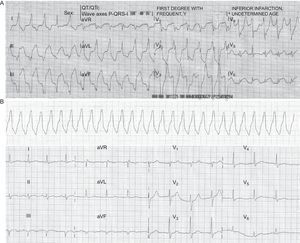
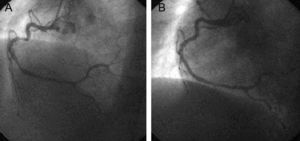
The patient was a 47-year-old male smoker, with type 2 diabetes without prior episodes of chest pain, who had experienced several syncope episodes at home. He had been treated by the emergency services, with documented SMVT at 140 bpm, with left bundle-branch block morphology and superior axis (Fig. 1A). Sinus rhythm was restored by electrical cardioversion. There was ST-segment elevation in the inferior and lateral wall leads with criteria for urgent revascularization; he was admitted to the intensive care unit of the referring hospital, where he underwent effective fibrinolysis. A few hours later he experienced a new syncopal SMVT at 170 bpm (telemetry recording alone, Fig. 1B) requiring electrical cardioversion. As this strongly suggested an acute adverse course the patient was referred to our center for urgent coronary angiography. Ventriculography showed depressed left ventricular systolic function (45%) with inferior akinesia, and coronary angiography showed diffuse disease with a large lesion with thrombotic material in the right coronary artery; this was treated by aspiration and the implantation of 2 bare-metal stents (Fig. 2). Outcome was good with Killip class I, decreased ST-segment elevation (without returning to a normal level) in the inferior wall leads (Fig. 1C) and a typical enzymatic curve with a peak troponin I value of 126 ng/mL. Seven days after the AMI, ventricular function was slightly depressed (left ventricular ejection fraction [LVEF] 45%-50% on endocardial enhanced visualization with cardiac magnetic resonance imaging), with no further SMVT episodes. Electrophysiological studies were conducted using the stimulation protocol developed by Josephson, and nonsustained syncopal ventricular flutter was induced by trains of stimuli and 2 extrastimuli at the right ventricular outflow tract. This was interpreted as being nonspecific and given that SMVT had been present in the acute phase of AMI, we decided to administer beta blockers at the maximum tolerated dose and maintain clinical monitoring.

A: 12-lead electrocardiogram during sustained monomorphic ventricular tachycardia at 140 bpm, with left bundle-branch block morphology, superior axis and fusion complexes. B: 1-lead telemetry tracing of sustained monomorphic ventricular tachycardia at 160 bpm. C: 12-lead electrocardiogram in sinus rhythm with a mild residual decrease in ST-segment elevation in inferior wall leads.

Nine months after discharge, the patient had experienced no further episodes of syncope or SMVT, and LVEF remained unchanged.
The presence of SMVT in the acute phase of AMI is rare and is usually associated with very extensive necrosis in an anterior location and congestive heart failure.1 In the study by Mont et al.,2 only 1.9% of patients with AMI showed SMVT in the first 48 h and the percentage is even lower (1.1%) if only an inferior location is taken into account. Interestingly, the factors that these authors identified as predisposing to SMVT (extensive necrosis, worse Killip class and bifascicular block) were not present in our patient. Given such an atypical clinical profile, it does not appear reasonable to expect similar results to those described regarding mortality (43%) and the recurrence of SMVT (17%), which may be closely related to ventricular dysfunction, nor does this profile appear to fit within the framework described in clinical practice guidelines.3 However, this atypical presentation should not modify the usual therapeutic strategy for urgent reperfusion and conventional medical treatment of AMI, nor does it entail indications for an implantable cardioverter defibrillator since arrhythmic events occur in the acute phase and LVEF is not very depressed.
The interest of this case lies in the patient's clinical characteristics and the diagnostic approach for subsequent management, since this was guided by the pathophysiological mechanism underlying the episodes of SMVT.
If it is thought that there is a previous anatomical substrate—whether idiopathic, or caused by a silent infarction (which is possible in a diabetic patient)—on which the ischemia has acted as a trigger, correcting the ischemia will not eliminate the risk of recurrent SMVT. The absence of inducibility does not rule out the presence of a substrate; however, because this may progress to transmural infarction, it does not appear reasonable to consider ablation (of the substrate), at least in the acute phase of AMI.4
However, if it is thought that the ventricular tachycardia episodes are secondary to a correctable cause, ischemia, which would have created a functional substrate different from the electrical instability that often causes far more severe polymorphic ventricular tachycardia or ventricular fibrillation (ischemia is not often a determining factor in the development of SMVT),5 then the principal treatment would be revascularization.
.
We would like to thank Dr Rafael Peinado Peinado for his contributions to the discussion.