The expression levels of microRNA-16-5p (miR-16) are upregulated in ischemic cardiomyopathy and in animal models of ischemic dilated cardiomyopathy (iDCM), inducing myocardial apoptosis. We investigated the role of miR-16 in the adaptive cellular response associated with endoplasmic reticulum (ER) stress and autophagy in the apoptotic iDCM environment.
MethodsWe quantified the miR-16 plasma levels of 168 participants—76 controls, 60 iDCM patients, and 32 familial DCM patients with the pathogenic variant of BAG3—by quantitative real-time polymerase chain reaction and correlated the levels with patient variables. The effects of intracellular miR-16 overexpression were analyzed in a human cardiac cell line. Apoptosis and cell viability were measured, as well as the levels of markers associated with ER stress, cardiac injury, and autophagy.
ResultsPlasma miR-16 levels were upregulated in iDCM patients (P=.039). A multivariate logistic regression model determined the association of miR-16 with iDCM clinical variables (P <.001). In vitro, miR-16 overexpression increased apoptosis (P=.02) and reduced cell viability (P=.008). Furthermore, it induced proapoptotic components of ER stress, based on upregulation of the PERK/CHOP pathway. However, we observed augmentation of autophagic flux (P <.001) without lysosomal blockade by miR-16 as a possible cytoprotective mechanism.
ConclusionsMiR-16 is specifically associated with iDCM. In an ischemic setting, miR-16 activates ER stress and promotes inflammation followed by autophagy in human cardiac cells. Thus, autophagy may be an attempt to maintain cellular homeostasis in response to misfolded/aggregated proteins related to ER stress, prior to apoptosis.
Keywords
Heart failure (HF) has reached pandemic proportions worldwide and is associated with high levels of morbidity and mortality.1 A common cause of HF is dilated cardiomyopathy (DCM).1 DCM is defined by left ventricular (LV) or biventricular dilation and impaired contraction that is not explained by abnormal loading conditions (ie, hypertension and valvular heart disease) or coronary artery disease.2 Ischemic DCM (iDCM) is the most frequent DCM etiology.3 It affects patients with a history of acute myocardial infarction or revascularization or is caused by coronary artery disease.4
The iDCM pathophysiology at subcellular levels remains unclear.5 After an ischemic event, apoptosis plays a crucial role in HF.6 Cardiomyocyte homeostasis involves multiple cellular processes, including endoplasmic reticulum (ER) stress and autophagy.7 ER stress signaling exerts a cytoprotective role and attempts to maintain this homeostasis but prolonged ER stress leads to cardiomyocyte dysfunction and apoptosis.7 On the other hand, autophagy vacuoles have been identified in the left ventricle of DCM patients.8,9 However, a deeper understanding is needed of the link between ER stress and autophagy in relation to cardiovascular diseases.
Circulating microRNAs (miRNAs) have gained prominence as diagnostic biomarkers and as a target for the treatment of DCM (extensively reviewed elsewhere).3 miRNAs play key roles in the complex signaling pathways underlying the pathophysiology of several cardiovascular diseases, including DCM. For example, microRNA-16-5p (miR-16) and miR-27 are positively related to LV impairment after acute myocardial infarction.10 Furthermore, the circulating and intracellular levels of miR-16 levels are upregulated in iDCM rat models11,12 and human fetal heart.13 miR-16 overexpression promotes myocardial apoptosis by regulating the expression of B-cell lymphoma 2 (BCL-2) in a rat ischemic model.12 In the same model, knockdown of miR-16 plays a cardioprotective role.11 However, the influence of miR-16 on intracellular mechanisms in the human iDCM population remains to be defined.
Our aim was to determine the role of miR-16 in the adaptive cellular response related to ER stress and autophagy under apoptotic iDCM conditions.
METHODSStudy population and blood samplingIn this case-control study, we enrolled 76 controls, 60 iDCM patients, and 32 familial DCM patients (with a pathogenic variant of BAG3). Only individuals older than 18 years were eligible. Detailed clinical and pharmacological information was obtained from each participant. The presence of iDCM was confirmed in all iDCM patients via coronary artery catheterization, as recommended by European Society of Cardiology guidelines.2 Plasma samples were obtained as previously described.14–16 Our protocol was approved by the Ethics Committee of Cádiz (Spain). All patients provided written informed consent. This study was conducted following the ethical principles of the Declaration of Helsinki.
Quantification of circulating miRNAsTotal RNA was extracted using the miRNeasy Serum/Plasma Advanced Kit (Qiagen, Germany). Circulating miR-16 levels were assessed by quantitative real-time polymerase chain reaction (qRT-PCR). miRNA-specific cDNA was synthesized with the mercury LNA RT kit (Qiagen). qRT-PCR was performed with the mercury SYBR Green PCR kit (Qiagen) and miR-16 levels were normalized by the 2−ΔCq method.
Cell culture and transfectionThe human AC16 cell line, which develops many of the biochemical properties of cardiac cells, was grown as previously described.17 Cells were transfected with 50pmol of hsa-miR-16-5p (miR-16) and positive (miR-1) and negative (Negative control #1) mirVana miRNA mimics using Lipofectamine RNAiMAX and following the manufacturer's recommendations (Thermo Fisher Scientific, United States) for 6, 16, 24, and 48hours. To measure the autophagic flux, the cells were treated with 2 autophagy control compounds—120μM chloroquine and 500nM rapamycin (Sigma-Aldrich, United States)—for 16 and 2hours, respectively.
Total RNA isolation and quantitative real-time polymerase chain reaction in AC16 cellsRNA from AC16 cells was isolated using the MagMAX Viral RNA Isolation kit (Thermo Fisher Scientific). RNA was reverse-transcribed using the High-Capacity cDNA Reverse Transcription kit (Thermo Fisher Scientific) following the manufacturer's instructions. miR-16, ATG14, TWF1, U6 snRNA, and GAPDH gene expression analysis was performed using a commercial TaqMan Gene Expression Assay (Applied Biosystems, United States). The relative quantification of mRNA levels (primer pairs are shown in table 1) was performed using 6.5 ng of cDNA, forward and reverse primers at 100nM each, and a SYBR Green PCR Master Mix Reagent kit (Life Technologies, United States). mRNA levels were normalized to those of GAPDH.
Oligonucleotides for quantitative real-time polymerase chain reaction
| Forward | Reverse | |
|---|---|---|
| ATF4 | 5′-TCTCCAGCGACAAGGCTAA-3’ | 5′-CCAATCTGTCCCGGAGAA-3’ |
| ATF6 | 5′-AATACTGAACTATGGACCTATGAGCA-3’ | 5′-TTGCAGGGCTCACACTAGG-3’ |
| ATG3 | 5′-CCTATTATGCCTAACTGGCACAT-3’ | 5′-TAGACAGTCTTCCAAGTTGCTACC-3’ |
| ATG4B | 5′-CAGATGATCTTTGCCCAAGC-3’ | 5′-TGGCTGCCTCTTCCTTTG-3’ |
| ATG14 | 5′-TGGGGACTACTCTGCCTACTACA-3’ | 5′-GGGTTACTCTGCTCCATGTCA-3’ |
| BCL-2 | 5′-AGCACGTGCACAGCTTCA-3’ | 5′-GTCCACGGGTGAAACAGC-3’ |
| proBNP | 5′-CGCAAAATGGTCCTCTACAC-3’ | 5′-CCGTGGAAATTTTGTGCTC-3’ |
| CASP3 | 5′-TGAGTGCTCGCAGCTCATA-3’ | 5′-GGGCTCGCTAACTCCTCAC-3’ |
| CHOP | 5′-TCACCACACCTGAAAGCAGA-3’ | 5′-TCTTGCAGGTCCTCATACCA-3’ |
| EDEM | 5′-CGGAAAGCGCTGGTAGAA-3’ | 5′-TGGAGTATTGTGGCTCGTCTT-3’ |
| GAPDH | 5′-AGCCACATCGCTCAGACAC-3’ | 5′-AATACGACCAAATCCGTTGACT-3’ |
| GRP78 | 5′-AATGACCAGAATCGCCTGAC-3’ | 5′-ATGCGCTCCTTGAGCTTTT-3’ |
| IL-1β | 5′GAAATCACACATGAACGTAGCC-3’ | 5′-CAAGTCATCCTCATTGCCACT-3’ |
| IRE1α | 5′-CCATCGAGCTGTGTGCAG-3’ | 5′-TGTTGAGGGAGTGGAGGTG-3’ |
| MAP1LC3B | 5′-GGTGTTGCCAGTTCCTGTCT-3’ | 5′-TCGCAAACCCAAGAGAAGTC-3’ |
| OS-9 | 5′-GGTCCAAGTGCGACCTTAAT-3’ | 5′-GTCCCCAGAGATACCTGCAC-3’ |
| PERK | 5′-TGGCCAGAAGTTGACAAAAGA-3’ | 5′-GGCAGCAATTCTCCCATCTA-3’ |
| P62 | 5′-ACATGGGGCTTGAGAAAGG-3’ | 5′-ATGCTACCGATGGTGAGGTC-3’ |
| TNNT2 | 5’-GGCTGCAGTGGCTACAGG-3’ | 5’CTGTCACCAGGCAATACAGC-3’ |
| ULK1 | 5′-CTGGCCGACTACCTGCAC-3’ | 5′-GGATGATGCCTTTGCTGTG-3’ |
| XBP1(s) | 5′-GCTGAGTCCGCAGCAGGT-3’ | 5′-GCTGAGTCCGCAGCAGGT-3’ |
Apoptosis was measured as the activation of caspase-3 and -7 using the FLICA assay (Immunochemistry Technologies, United States) following the manufacturer's instructions.
Cell viability assayCell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Sigma-Aldrich) as previously described.18
Autophagy assayThe CYTO-ID Autophagy Detection Kit (Enzo Life Sciences, United States) was used to measure autophagosome formation following the manufacturer's instructions.
Western blot analysisTransfected AC16 cells were harvested in RIPA buffer (Sigma-Aldrich), and protein concentrations were determined using a BCA protein assay kit (Thermo Fisher Scientific). The primary antibodies used for western blotting were anti-caspase-3, anti-cleaved-caspase-3, anti-GRP78, anti-LC3-B, anti-P62 (all from Cell Signaling Technology, Netherlands), anti-ATG14 (Abcam, United Kingdom), and β-actin (Sigma-Aldrich). Protein bands were visualized using the ECL immunoblotting detection system (Bio-Rad, United States) and developed on a ChemiDoc MP (Bio-Rad) imaging system. To analyze protein expression, bands were quantified by densitometry using Image Lab analysis software (Bio-Rad).
Statistical analysisData are expressed as the mean±standard error of the mean (SEM) unless otherwise described. Statistical significance was defined as P <.05. Analysis of differences among healthy, iDCM, and familial DCM groups was performed using analysis of variance (ANOVA). Receiver operator characteristic (ROC) curves to characterize the diagnostic performance of miR-16 were plotted to determine the area under the curve (AUC), specificity, and sensitivity of the optimal cutoffs. ROC curves were generated by plotting sensitivity against 100−specificity. Data are presented as the AUC and 95% confidence interval (95%CI). Student t test was used to analyze differences after miR-16 overexpression in AC16 cells. Relationships between miRNAs and LV ejection fraction were assessed using logistic regression. The Wilcoxon test and iterating combinations between miR-16, besides echocardiographic and clinical covariates, were used to construct several models. Changes in the P values of their variables were evaluated by the Wald test and a likelihood ratio. The statistical package R was used for all analyses. Figures were generated using GraphPad Prism 6 software.
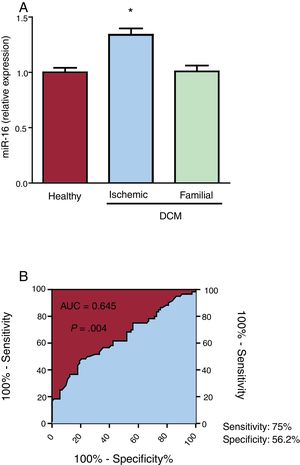
RESULTSmiR-16 is upregulated in the plasma of ischemic dilated cardiomyopathy patientsPatient characteristics are listed in table 2. miR-16 expression was 1.34-fold higher in iDCM patient plasma than in that of healthy controls (P=.039; figure 1A). Interestingly, no differences were observed in miR-16 plasma levels between familial DCM patients and healthy patients. ROC analysis of miR-16 showed a significant AUC (figure 1B). The discriminatory power to distinguish iDCM patients from controls was 0.645 (95%CI, 0.55-0.74; P=.004). This cutoff gave a sensitivity of 75.0% and specificity of 56.2% for the identification of iDCM patients and healthy controls.
Demographic, clinical, and echocardiographic variables of the study population
| Variable | Healthy control (n=76) | Ischemic DCM (n=60) | Familial DCM (n=32) |
|---|---|---|---|
| Age, y | 38.3±11.8 | 68.2±8.3 | 42.3±15.3 |
| Male sex | 38 (52.1) | 48 (80) | 21 (65.6) |
| BMI, kg/m2 | 23.8±3.5 | 28.8±4.3 | 27.6±4.8 |
| Current smoker | - | 31 (59) | 8 (25) |
| LVEF, % | 66.4±5.2 | 34.1±6.0 | 47.3±11.7 |
| LVEDD, mm | 45.6±4.9 | 61.0±6.0 | 56.5±8.4 |
| LVESD, mm | 28.1±5.1 | 45.3±15.0 | 42.2±10.8 |
| LAD, mm | 36.4±8.3 | 44.5±7.6 | 38.2±6.5 |
| Sphericity index | 0.5±0.4 | 0.7±0.1 | 0.8±0.2 |
| TDI s’, cm/s | 0.10±0.02 | 0.05±0.01 | 0.09±0.02 |
| E/e’ ratio | 7.1±1.8 | 15.5±7.1 | 7.6±2.6 |
| NYHA functional class (II-III) | - | 68 | 34 |
| ARBs | - | 29 (48.3) | 6 (18.8) |
| ACE inhibitors | - | 24 (40) | 10 (31.6) |
| Nitrates | - | 14 (23.3) | - |
| MRAs | - | 38 (63.3) | 8 (25) |
| Antiplatelet agents | - | 36 (60) | 2 (6.3) |
| Statins | 28 (36.8) | 19 (31.7) | 3 (9.4) |
| Beta-blockers | - | 50 (83.3) | 4 (12.5) |
ACE inhibitors, angiotensin-converting enzyme inhibitors; ARBs, angiotensin II receptor blockers; BMI, body mass index; DCM, dilated cardiomyopathy; LAD, left atrial dimension; LVEDD, left ventricular end-diastolic dimension; LVEF, left ventricle ejection fraction; LVESD, left ventricle end-systolic dimension; MRAs, aldosterone receptor antagonists; NYHA, New York Heart Association; TDI s’, tissue Doppler imaging peak systolic wave.
All values are expressed as mean±standard error of the mean or No. (percentage).
 expression detected by quantitative real-time polymerase chain reaction (qRT-PCR) in healthy individuals and in ischemic and familial dilated cardiomyopathy (DCM) patients. A: miR-16 expression was significantly increased in ischemic DCM patients (n=60) compared with familial DCM patients (n=32) and healthy individuals (n=76). B: receiver operating characteristic (ROC) analysis of miR-16 for ischemic DCM, with an area under curve (AUC) of 0.645 (95%CI, 0.55-0.74; P=.004), sensitivity of 75%, and specificity of 56.2%. *P <.05.")
Plasma microRNA-16-5p (miR-16) expression detected by quantitative real-time polymerase chain reaction (qRT-PCR) in healthy individuals and in ischemic and familial dilated cardiomyopathy (DCM) patients. A: miR-16 expression was significantly increased in ischemic DCM patients (n=60) compared with familial DCM patients (n=32) and healthy individuals (n=76). B: receiver operating characteristic (ROC) analysis of miR-16 for ischemic DCM, with an area under curve (AUC) of 0.645 (95%CI, 0.55-0.74; P=.004), sensitivity of 75%, and specificity of 56.2%. *P <.05.
Multivariate logistic regression analyses showed that circulating miR-16 levels were associated with the sphericity index and tissue Doppler imaging systolic peak wave (TDI s’). Based on this analysis, the sphericity index (odds ratio [OR], 1.20; 95%CI, 1.07-1.34; P <.001), TDI s’ (OR, 0.44; 95%CI, 0.30-0.65; P <.001), and plasma miR-16 level (OR, 8.53; 95%CI, 1.30-55.82; P=.013) were independent factors for iDCM (table 3). The Akaike Information Criterion showed a value of 62.15. ROC analysis of the multivariate model showed an AUC of 0.963 (95%CI, 0.92-1.00; P <.001).
Multivariate logistic regression analysis of iDCM patients
| Variable | OR | 95%CI | P |
|---|---|---|---|
| Sphericity index | 1.20 | 1.07-1.34 | <.001 |
| TDI s’ | 0.44 | 0.30-0.65 | <.001 |
| miR-16 | 8.53 | 1.30-55.82 | .013 |
| AIC value: 62.15 |
95%CI, 95% confidence interval; AIC, Akaike information criterion; iDCM, ischemic dilated cardiomyopathy; OR, odds ratio; TDI s’, tissue Doppler imaging systolic peak wave.
The model included the sphericity index, TDI s’, and the plasma level of miR-16.
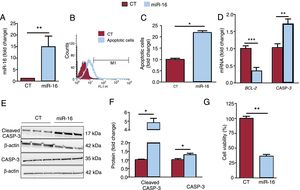
miR-16 levels were upregulated (figure 2A), and this overexpression induced apoptosis (2.187-fold change in apoptotic cells, P=.02) (figure 2B,C). These results are consistent with the lower mRNA levels of BCL-2 (0.35-fold change, P=.0007) (figure 2D) and higher protein levels of cleaved caspase-3 (4.82-fold change, P=.02) (figure 2E,F). Furthermore, miR-16 reduced cell viability in miR-16-overexpressing AC16 cells (a 64% reduction; P=.008) (figure 2G).
 promotes apoptosis in human cardiac cells. A: the relative mRNA expression of miR-16 was detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of U6 SnRNA (n=5). B: the presence of apoptotic cells was determined using the FLICA assay by flow cytometry. C: quantitative flow cytometry results of apoptotic cardiac cells are shown (n=3). D: relative mRNA levels of BCL-2 and caspase-3 mRNA were detected by qRT-PCR and normalized to that of GAPDH (n=7). E: whole cell lysates were subjected to immunoblot analysis with specific antibodies against cleaved caspase-3, caspase-3, and β-actin (n=3). F: quantification of cleaved caspase-3 and caspase-3 levels, normalized to that of β-actin (n=3). G: cell viability was assessed by MTT assay; data are presented as % of cell viability. Results are expressed as mean±standard error of the mean. CASP-3, caspase-3; CT, control AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells. *P < .05, **P < .01, ***P < .005.")
Overexpression of microRNA-16-5p (miR-16) promotes apoptosis in human cardiac cells. A: the relative mRNA expression of miR-16 was detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of U6 SnRNA (n=5). B: the presence of apoptotic cells was determined using the FLICA assay by flow cytometry. C: quantitative flow cytometry results of apoptotic cardiac cells are shown (n=3). D: relative mRNA levels of BCL-2 and caspase-3 mRNA were detected by qRT-PCR and normalized to that of GAPDH (n=7). E: whole cell lysates were subjected to immunoblot analysis with specific antibodies against cleaved caspase-3, caspase-3, and β-actin (n=3). F: quantification of cleaved caspase-3 and caspase-3 levels, normalized to that of β-actin (n=3). G: cell viability was assessed by MTT assay; data are presented as % of cell viability. Results are expressed as mean±standard error of the mean. CASP-3, caspase-3; CT, control AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells. *P < .05, **P < .01, ***P < .005.
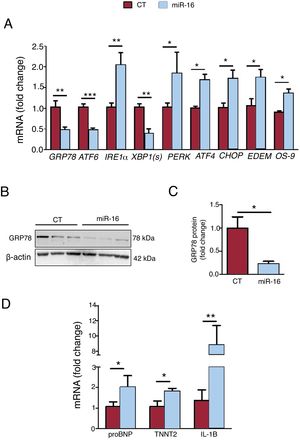
Next, we analyzed the levels of key genes involved in ER stress, the ER-associated protein degradation (ERAD) response, and HF biomarkers (figure 3A-C). miR-16 overexpression promoted the pro-apoptotic ER stress cascade, based on the mRNA expression levels of the PERK/CHOP pathway: PERK (1.84-fold change, P=.01), ATF4 (1.68-fold change, P=.02), and CHOP (1.72-fold change, P=.03) (figure 3A). On the other hand, we observed a fall in GRP78 (mRNA: 0.47-fold change, P=.004; protein: 0.22-fold change, P=.04) and in the mRNA levels of ATF6 (0.48-fold change, P=.002) and XBP1(s) (0.38-fold change, P=.01). In addition, miR-16 overexpression activated the ERAD response (EDEM mRNA: 1.74-fold change, P=.03; OS-9 mRNA: 1.36-fold change, P=.02). Regarding HF biomarkers, miR-16 overexpression promoted the expression of pro-B-type natriuretic peptide (proBNP) (2.03-fold change, P=.03) and cardiac muscle troponin T (TNNT2) (1.83-fold change, P=.02) (figure 3C). Moreover, IL-1β mRNA levels were 8.84-fold higher (P=.035) (figure 3C).
 on endoplasmic reticulum (ER) stress, heart failure biomarkers, and inflammation in human cardiac cells. A: relative mRNA levels of ER stress markers (GRP78, ATF6, IRE1α, XBP1(s), ATF4, CHOP, EDEM, and OS-9) were detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of GAPDH (n=5). B: whole cell lysates were subjected to immunoblot analysis with specific antibodies against GRP78 and β-actin (n=3). C: quantification of GRP78 protein levels, normalized to that of β-actin (n=3). D: relative mRNA levels of HF (pro-B-type natriuretic peptide and TNNT2) and inflammatory (IL-1B) biomarkers were detected by qRT-PCR and normalized to that of GAPDH (n=5). Results are expressed as mean±standard error of the mean. CT, control AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells. *P < .05, **P < .005.")
Effects of microRNA-16-5p (miR-16) on endoplasmic reticulum (ER) stress, heart failure biomarkers, and inflammation in human cardiac cells. A: relative mRNA levels of ER stress markers (GRP78, ATF6, IRE1α, XBP1(s), ATF4, CHOP, EDEM, and OS-9) were detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of GAPDH (n=5). B: whole cell lysates were subjected to immunoblot analysis with specific antibodies against GRP78 and β-actin (n=3). C: quantification of GRP78 protein levels, normalized to that of β-actin (n=3). D: relative mRNA levels of HF (pro-B-type natriuretic peptide and TNNT2) and inflammatory (IL-1B) biomarkers were detected by qRT-PCR and normalized to that of GAPDH (n=5). Results are expressed as mean±standard error of the mean. CT, control AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells. *P < .05, **P < .005.
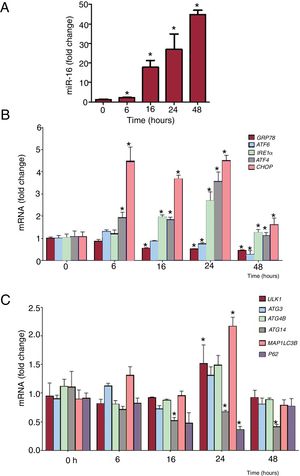
Time-course experiments were then performed to further characterize the ER stress activation induced by miR-16 (figure 4). The mRNA levels of IRE1α (1.84-fold change, P=.02), ATF4 (1.96-fold change, P=.03), and CHOP (3.68-fold change, P=.002) were significantly increased 6 to 16hours after miR-16 transfection. The mRNA levels of ATF6 and GRP78 were decreased in response to miR-16 overexpression (figure 4A,B). Autophagy-related transcripts were next analyzed. The mRNA levels of ULK1 (1.57-fold change, P=.04) and MAP1LC3B (2.17-fold change, P=.04) were significantly increased 24hours after miR-16 transfection. The mRNA levels of ATG14 were decreased under miR-16 overexpression conditions.
 overexpression promotes endoplasmic reticulum (ER) stress prior to autophagy in human cardiac cells. A: relative miR-16 levels were detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of U6 SnRNA (n=4). B: relative mRNA levels of ER stress markers (GRP78, ATF6, IRE1α, ATF4, and CHOP) were detected by qRT-PCR and normalized to that of GAPDH (n=4). C: relative mRNA levels of autophagy markers (ULK1, ATG3, ATG4B, ATG14, MAP1LC3B, and p62) were detected by qRT-PCR and normalized to that of GAPDH (n=4). Results are expressed as mean±standard error of the mean. *P <.05.")
MicroRNA-16-5p (miR-16) overexpression promotes endoplasmic reticulum (ER) stress prior to autophagy in human cardiac cells. A: relative miR-16 levels were detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of U6 SnRNA (n=4). B: relative mRNA levels of ER stress markers (GRP78, ATF6, IRE1α, ATF4, and CHOP) were detected by qRT-PCR and normalized to that of GAPDH (n=4). C: relative mRNA levels of autophagy markers (ULK1, ATG3, ATG4B, ATG14, MAP1LC3B, and p62) were detected by qRT-PCR and normalized to that of GAPDH (n=4). Results are expressed as mean±standard error of the mean. *P <.05.
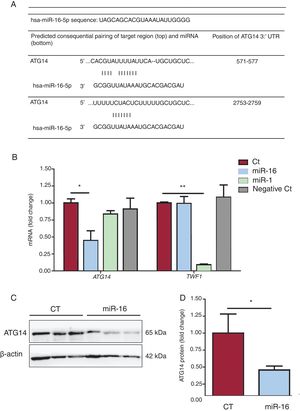
Bioinformatic prediction tools (TargetScan, miRBase, and miRDB)19 revealed the binding sequence of the miRNA–mRNA interaction (figure 5A). ATG14 and TWF1 mRNA expression levels were reduced 0.448- and 0.091-fold after miR-16 and miR-1 overexpression, respectively (figure 5B). These results were corroborated by the reduction in ATG14 protein levels compared with controls (0.16-fold change, P=.03) (figure 6C), which further confirmed that miR-16 can negatively regulate ATG14 expression in AC16 cells.
 and normalized to that of GAPDH (n=4). C: whole cell lysates were subjected to immunoblot analysis with specific antibodies against ATG14 and β-actin. D: quantification of ATG14 levels, normalized to that of β-actin (n=3). Results are expressed as mean±standard error of the mean. CT, control AC16 cells; miR-1, microRNA-1–positive control-overexpressing AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells; Negative Ct, negative control AC16 cells. *P < .05, **P < .005.")
Analysis of ATG14 gene expression. A: pairing of target Atg14 regions and miR-16 predicted by TargetScan, miRBase, and miRDB. B: relative mRNA levels of ATG14 and TWF1 were detected by quantitative real-time polymerase chain reaction (qRT-PCR) and normalized to that of GAPDH (n=4). C: whole cell lysates were subjected to immunoblot analysis with specific antibodies against ATG14 and β-actin. D: quantification of ATG14 levels, normalized to that of β-actin (n=3). Results are expressed as mean±standard error of the mean. CT, control AC16 cells; miR-1, microRNA-1–positive control-overexpressing AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells; Negative Ct, negative control AC16 cells. *P < .05, **P < .005.
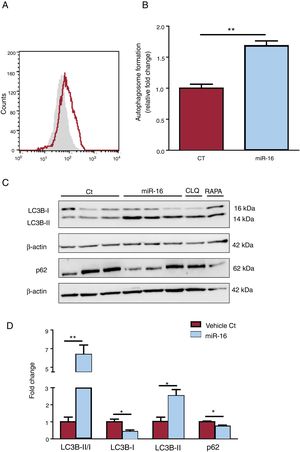
 on autophagy in human cardiac cells. A: the presence of autophagosomes was determined using CYTO-ID and flow cytometry. B: quantitative flow cytometry results are shown (n=3). C: whole cell lysates were subjected to immunoblot analysis with specific antibodies against LC3B (I and II), p62, and β-actin (n=3). D: quantification of the LC3BII/I ratio and p62 levels, both normalized to that of β-actin (n=3). Results are expressed as mean±standard error of the mean. n=3 or 4. CT, control AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells. *P < .05, **P < .005.")
Effects of microRNA-16-5p (miR-16) on autophagy in human cardiac cells. A: the presence of autophagosomes was determined using CYTO-ID and flow cytometry. B: quantitative flow cytometry results are shown (n=3). C: whole cell lysates were subjected to immunoblot analysis with specific antibodies against LC3B (I and II), p62, and β-actin (n=3). D: quantification of the LC3BII/I ratio and p62 levels, both normalized to that of β-actin (n=3). Results are expressed as mean±standard error of the mean. n=3 or 4. CT, control AC16 cells; miR-16, microRNA-16-5p–overexpressing AC16 cells. *P < .05, **P < .005.
miR-16 overexpression leads to autophagosome accumulation, as shown by an increase in CYTO-ID dye intensity compared with controls (1.69-fold change, P <.001) (figure 6A,B). The LC3BI/II ratio and p62 were assessed as representative proteins of the autophagic flux (figure 6C,D). A significant increase in conversion from LC3B-I to LC3B-II (6.37-fold change, P=.003) and p62 protein degradation (0.74-fold change, P=.01) reflected autophagic activity in miR-16–overexpressing cardiac cells.
DISCUSSIONDCM is the most common cardiomyopathy and the main reason for heart transplantation worldwide.20 It is a complex disease with a common phenotype encompassing a heterogeneous etiological mechanism. Improvements in the pathophysiology underlying DCM could reduce the clinical burden of this entity. Hence, novel noninvasive indicators, such as miRNA, are still needed in clinical decision-making and in the development of therapeutic strategies. Furthermore, identification of anomalous miRNAs and their targets may reveal the underlying pathogenesis of iDCM at the molecular level.3,21–26 We found that miR-16 expression was significantly higher in the plasma of iDCM patients than in that of healthy and familial DCM individuals, confirming that miR-16 is related to iDCM. Moreover, various echocardiographic parameters such as sphericity index and peak systolic TDI s’ showed an association with the iDCM cohort. A lower sphericity index and TDI s’ have been linked to worse prognosis in DCM. Together with the results obtained by Devaux et al.,10 we propose a multiparametric model including sphericity index, TDI s’, and miR-16 that may improve the identification of iDCM. Therefore, miR-16 shows promise as an iDCM biomarker for possible clinical translation.
miR-16 overexpression inhibits cell proliferation and promotes apoptosis by directly targeting BCL-2 in rat ischemic models.11,12 In our study, we mimicked this situation in a human cardiac cell line in terms of apoptosis, cell viability, and BCL-2. Conversely, it has been described that BCL-2 levels are increased and may be involved in a compensatory mechanism in the left ventricle of the DCM population.24 On the other hand, cell death is activated in the left ventricle of patients with end-stage DCM.27 Liu et al.27 reported that caspase-3 and CHOP, key proteins involved in cell death, were activated in LV tissues from end-stage DCM patients compared with normal heart tissue. Our data demonstrated that miR-16 overexpression promotes the upregulation of CHOP mRNA and of the protein levels of caspase-3 and cleaved caspase-3. Nevertheless, more evidence is required to determine the successive pathological events that converge in cardiomyocyte apoptosis.
BCL-2, one of the essential antiapoptotic proteins, plays a central role in adaptation to cellular stress, including the molecular crosstalk between ER stress and autophagy.28,29 Because BCL-2 is a main target of miR-16, these 2 adaptive cellular responses were analyzed. Several findings suggest that the proapoptotic sequence related to ER stress is influenced by miR-16 in human cardiac cells. First, overexpression of CHOP leads to cell apoptosis and cell cycle arrest.30,31 Our results confirmed that CHOP is upregulated soon after miR-16 overexpression, with a subsequent increase in the number of apoptotic cardiac cells. CHOP regulates several pro- and antiapoptotic genes, including caspase-3 and BCL-2 transcription.32 Under ER stress conditions, BCL-2 expression is downregulated by CHOP, which sensitizes cells to apoptosis.33 Second, the levels of GRP78, a master cytoprotective regulator that reduces ER stress levels, are increased after ischemic simulation in a rat cardiomyocyte model.34 However, miR-16 overexpression in our cell model reduced GRP78 levels. This may be due to downregulation of ATF6, targeted by miR-16,35 or to the general inhibition of protein translation by the PERK/CHOP pathway.
Overwhelming ER stress promotes inflammation and HF.7,36 ProBNP, the prohormone form of the natriuretic peptides (BNP and N-terminal proBNP), and TNNT2 are significant clinical biomarkers for the diagnosis of cardiac dysfunction after an ischemic event.37,38 We confirmed that miR-16 leads to the expression of proBNP and TNNT2. Under pathological conditions (ie, mechanical stress or ischemia), LV cells express proBNP as a response to volume expansion of the ventricle.37 TNNT2 is an element of the contractile apparatus of cardiomyocytes. It regulates muscle contraction in response to alterations in intracellular calcium and indicates highly specific cardiac muscle damage.38 Regarding inflammation, proinflammatory cytokines such as IL-1β increase the permeability of the cardiomyocyte cell membrane, promoting apoptosis and troponin release.39 Miyazaki et al.40 demonstrated that the PERK/CHOP-mediated pathway increases tissue inflammation, including IL-1β levels, after myocardial ischemia/reperfusion injury in a mouse model. miR-16 overexpression leads to the upregulation of IL-1β mRNA. In addition, the PERK/CHOP pathway plays a specific role in controlling IL-1β production and maturation.41 Moreover, IL-1β levels can predict mortality and the need for heart transplantation in DCM patients.42
ER stress and inflammation may initiate the process of autophagy in the pathogenesis of heart diseases.7,43 Our data demonstrate that autophagy is activated after ER stress, possibly as a response to misfolded/aggregated proteins produced by miR-16 overexpression. The presence of autophagosomes in the idiopathic DCM population suggests improved HF prognosis. These results indicate a protective role of autophagy in myocardial impairment.8,9 We confirmed that miR-16 promotes autophagosome accumulation. More than 30 autophagy-related gene (ATG) proteins are involved in autophagy, particularly in autophagosome formation,29 with ATG14 one of the main proteins involved.44 Furthermore, ATG14 is one of the target genes regulated by miR-16 in vascular endothelial cells.45 In the present study, miR-16 inhibited autophagy progression and played a role in the development of vascular endothelial injury and chronic heart disease.45 ATG14 was significantly downregulated in our model after miR-16 overexpression. However, the autophagic flux was activated, which indicates that ATG14 might not play a principal role in this process. Recently, Caragnano et al.46 stated that the autophagic process is blocked in DCM patients, based on p62 protein accumulation. Hence, the role of autophagy as a pro-death or cytoprotective program remains undetermined in the various DCM etiologies. We propose that miR-16 promotes cell autophagy, reducing the levels of protein aggregates, as the final weapon to cope with the problem. Indeed, chronic ischemia has been described to activate autophagy47 and its stimulation protects cardiomyocytes from ischemic damage.48 However, the overwhelming upregulation of autophagy could result in cardiomyocyte apoptosis and worsen cardiac performance.43
In summary, our results confirm that miR-16 plays a significant role in the progression of human cardiac cell injury in iDCM. By acting through several molecular targets, miR-16 has a sequential impact on ER stress, inflammation, autophagy, and apoptosis. Hence, an improved understanding of the crosstalk between these processes and clarification of the role of miR-16 will boost the development of alternative and individualized therapeutic strategies for iDCM.
LimitationsThere are some limitations to this study. First, the usefulness of miR-16 should be compared with that of established biochemical tests. Second, due to the lack of heart biopsy samples, it was impossible to determine whether the elevated miR-16 plasma levels reflected the intracellular miRNA expression pattern. Although the use of a stable human cardiac cell line has advantages and is essential to describe miR-16 overexpression-related cellular and molecular processes, the clinical translation of these results requires further research. Although ATG14 expression was downregulated after miR-16 overexpression, a luciferase assay should be performed to determine whether ATG14 is directly targeted by miR-16 in this human cardiac cell line.
CONCLUSIONSIn conclusion, the plasma levels of miR-16 are upregulated in iDCM patients, confirming its relationship with this etiology. Our study reveals that miR-16 activates ER stress and promotes inflammation followed by autophagy in human cardiac cells under ischemic conditions. Thus, autophagy may be an attempt to maintain cellular homeostasis in response to misfolded/aggregated proteins related to ER stress, prior to apoptosis.
FUNDINGThis work was supported by grants in the framework of the Integrated Territorial Initiative (ITI PI0048-2017 and ITI0033_2019), FPS00136-2018, and a clinical research grant from the Spanish Society of Cardiology for Basic Research in Cardiology (PI0012_2019).
CONFLICTS OF INTERESTR. Toro and A. Mangas have filed a patent on the use of microRNAs as biomarkers. The other authors have nothing to declare.
- -
Despite being one of the main causes of HF, the pathophysiology of iDCM at the subcellular level remains unclear.
- -
There is growing evidence of the involvement of miRNA dysregulation in several cardiovascular diseases, including DCM.
- -
miR-16 levels are increased in iDCM rat models and iDCM patients.
- -
Its overexpression triggers myocardial apoptosis via the posttranscriptional regulation of BCL-2 in a rat cardiomyocyte ischemia model.
- -
Moreover, its absence from the cardiomyocyte improves cardiac function.
- -
Circulating miR-16 levels were significantly higher in the plasma of iDCM patients than in that of healthy controls and its levels were not increased in familial DCM patients, suggesting that miR-16 is specifically related to iDCM.
- -
Furthermore, our results indicate that miR-16 induces ER stress, HF markers, inflammation, autophagy, and apoptosis in human cardiac cells.
- -
An improved understanding of these processes and clarification of the role of miR-16 will boost the development of new therapeutic strategies for iDCM.
We thank all of the clinical staff who participated in the study and Lucille Banham for valuable assistance in the preparation of the English manuscript.