
Ivabradine reduces heart rate by blocking the I(f) current and preserves blood pressure and stroke volume through unknown mechanisms. Caveolin-3 protects the heart by forming protein complexes with several proteins, including extracellular matrix (ECM)-metalloproteinase-inducer (EMMPRIN) and hyperpolarization-activated cyclic nucleotide-gated channel 4 (HN4), a target of ivabradine. We hypothesized that ivabradine might also exert cardioprotective effects through inhibition of ECM degradation.
MethodsIn a porcine model of cardiogenic shock, we studied the effects of ivabradine on heart integrity, the levels of MMP-9 and EMMPRIN, and the stability of caveolin-3/HCN4 protein complexes with EMMPRIN.
ResultsAdministration of 0.3 mg/kg ivabradine significantly reduced cardiogenic shock-induced ventricular necrosis and expression of MMP-9 without affecting EMMPRIN mRNA, protein, or protein glycosylation (required for MMP activation). However, ivabradine increased the levels of the caveolin-3/LG-EMMPRIN (low-glycosylated EMMPRIN) and caveolin-3/HCN4 protein complexes and decreased that of a new complex between HCN4 and high-glycosylated EMMPRIN formed in response to cardiogenic shock. We next tested whether caveolin-3 can bind to HCN4 and EMMPRIN and found that the HCN4/EMMPRIN complex was preserved when we silenced caveolin-3 expression, indicating a direct interaction between these 2 proteins. Similarly, EMMPRIN-silenced cells showed a significant reduction in the binding of caveolin-3/HCN4, which regulates the I(f) current, suggesting that, rather than a direct interaction, both proteins bind to EMMPRIN.
ConclusionsIn addition to inhibition of the I(f) current, ivabradine may induce cardiac protection by inhibiting ECM degradation through preservation of the caveolin-3/LG-EMMPRIN complex and control heart rate by stabilizing the caveolin-3/HCN4 complex.
Keywords
Beta-blockers and calcium channel inhibitors have been prescribed in the last 20 years to reduce heart rate in patients with chronic heart failure.1 However, the development of new drugs was required for several reasons, including patient intolerance of beta-blocker administration and the negative inotropic effects of the drugs.
Ivabradine is a selective inhibitor of the pacemaker I(f) current in sinoatrial cells. By inhibiting hyperpolarization-activated cyclic nucleotide-gated channels 1 and 4 (HCN1 and -4), ivabradine reduces heart rate while preserving blood pressure and left ventricular ejection fraction (LVEF),2 without adverse effects. The open state of HCN channels, controlled by the intracellular concentration of cyclic adenosine monophosphate (cAMP), regulates diastolic depolarization. While beta-blocker administration significantly reduces cAMP-dependent heart rate, ivabradine maintains cardiac contractility regardless of heart rate,3 which makes this drug one of the most recommended in the treatment of chronic heart failure and left ventricular ejection fraction <35%. Nonetheless, its effectiveness for acute heart failure remains unclear.
Cardiogenic shock (CS) is the leading recurrent cause of death after acute myocardial infarction. Administration of vasoactive drugs in this context is necessary for adequate organ perfusion but may contribute to myocardial damage through extensive tachycardia, decreased ventricular efficiency, increased oxidative stress, and excessive myocardial demand for oxygen. We and others4 have found that ivabradine ameliorates catecholamine-induced sinus tachycardia in CS, improving hemodynamic parameters via unclear mechanisms.
A role for ivabradine in the preservation of the extracellular matrix (ECM) has recently been suggested in the context of osteoarthritis, diabetes,5 and atherosclerosis.6 Furthermore, ivabradine can reduce apoptosis in cardiac myocytes in chronic viral myocarditis,7 in the aortic constriction mouse model of cardiac hypertrophy,8 in acute and chronic heart failure,9,10 and in diabetic cardiomyopathy.11 However, how ivabradine helps to inhibit the degradation of the ECM and reduce cell death remains unknown.
Although HCN4 is the main target of ivabradine, it is unclear how HCN4 could alter ECM degradation. HCN4 forms macromolecular complexes with caveolin-3, a protein mainly expressed in the caveolae of cardiac myocytes, and this interaction is proposed to regulate HCN4 cardiac pacemaker activity.9 Likewise, caveolin-3 can interact with other proteins to modulate their function, as we have previously described in the setting of cardiac ischemia/reperfusion, in which caveolin-3 plays a cardioprotective role by binding to extracellular matrix metalloprotease inducer (EMMPRIN). This interaction inhibits not only the degradation of the ECM, but also cardiac apoptosis.8
To evaluate whether ivabradine can preserve myocardial contractility beyond the I(f) current, we studied the integrity of the heart in a porcine model of CS and analyzed the contribution of ivabradine to the preservation of the ECM by examining the enzymes involved in ECM degradation and the underlying molecular mechanisms.
METHODSAll procedures were performed in the experimental surgery facilities of our institute. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1985) and the Animal Welfare Ethics Committee, complied with the EU Directive on experimental animals (63/2010 EU) and related Spanish legislation (RD 53/2013), and received the relevant local approval (PROEX 365-15). Detailed procedures are presented in .
ReagentsA detailed list of reagents is presented in the .
Animals and cardiogenic shock procedureCS was induced in 20 Yorkshire female pigs (37.8±5.2kg); the survival rate was 90%. At the end of the experimental procedure, 10 animals received 0.3mg/kg ivabradine and 10 received placebo (saline). Two animals (1 from each group) did not survive beyond 24hours after the CS.
Pigs were premedicated with intramuscular ketamine (10mg/kg) and midazolam (0.5mg/kg). Anesthesia was induced by isoflurane inhalation and maintained with continuous infusion of propofol (2mL/kg/h), fentanyl (50mg/kg/h), and diazepam (10mg/kg/h). Animals were intubated and ventilated with 100% oxygen saturation. The animals received 5000 IU of heparin and amiodarone (2mg/kg/h) to avoid blood clotting in catheters and malignant cardiac arrhythmias, respectively.
Ischemia/reperfusion was induced by left anterior descending artery occlusion for 45minutes using a JL 3 6-Fr catheter and balloon inflation at 5 atmospheres. Volume overload was induced by administration of 500mL hydroxyethyl starch and 1500mL of 0.9% saline serum. Hemodynamic measurements were recorded at 15, 30, and 45minutes of ischemia. When ventricular fibrillation/ventricular tachycardia occurred, we administered a biphasic DC shock (10-20J) combined with direct manual chest compressions. Noradrenaline (20-80μg/kg/h), dobutamine (240-660μg/kg/h), and physiological saline (1000-2000mL) were administered until the heart rate exceeded 90 bpm. After 45minutes of left anterior descending artery occlusion, the animals were then randomized to a control or ivabradine group.
Ivabradine powder was weighed on a high-precision laboratory scale and diluted to at least 12mg/mL in distilled water. The solution was then transferred to a 10-mL syringe for slow-bolus intravenous administration to the animal at a dose of 0.3mL/kg. The control group received an equivalent volume of physiological saline.
Evans blue/TTC stainingMyocardial infarction extension was evaluated by 5% Evans blue perfusion (in serum) and TTC staining as described previously12 (for details, see the ).
Determination of troponin I plasma levelsPlasma troponin I was determined with the commercial Human Cardiac Troponin 1 SimpleStep ELISA kit (Abcam, Spain) following the manufacturer's instructions.
Cardiac ultrasoundPig hearts were visualized by echocardiography using a Vivid Q ultrasound system from GE Healthcare (United States) equipped with a 1.9- to 4.0-MHz scan head, as described previously.13 Further information is provided in the .
CellsH9c2 and HL1B cells were grown as described previously14 and in the . Cells were cultured under hypoxic conditions in hypoxia incubator chambers in a humidified atmosphere containing 1% oxygen, 5% carbon dioxide, and 94% nitrogen. Reoxygenation was performed with fresh medium.
Histology and immunohistochemistryHistological, immunohistochemical, and immunohistofluorescence procedures were performed as previously described.13,14
Immunoprecipitation and immunoblot analysisIsolation of protein lysates and immunoprecipitations and immunoblots were performed as described.13
RNA isolation and quantitative real-time polymerase chain reactionReal-time polymerase chain reaction (RT-PCR) was performed as previously described.8 Additional information is provided in the . The following primers were used: EMMPRIN-Forward: 5’-GGC ACC ATC GTA ACC TCT GT-3’; EMMPRIN-Reverse: 5’-CAC TGG CGT GTT CCG ATT TC-3’; β-actin-Forward: 5’-CTT AGT TGC GTT ACA CCC TTT CT-3’; β-actin-Reverse: 5’-CTG TCA CCT TCA CCG TTC CAG TT-3’.
Statistical analysisAll data were analyzed using SPSS 22.0 statistical software package (SPSS Inc, United States). All values are given as mean±standard deviation. Significance is reported at the 5% level. Whenever comparisons were made with a common control, the significance of differences was tested by analysis of variance followed by Dunnett's modification of the t test.
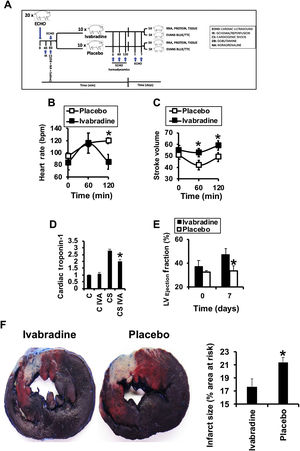
RESULTSIvabradine reduces left ventricular necrosis after cardiogenic shockIn pigs subjected to CS (figure 1A), intravenous administration of 0.3mg/kg ivabradine or saline was associated with a significant reduction in heart rate (figure 1B) but preserved stroke volume (figure 1C). The levels of cardiac troponin, indicative of ischemic injury, were reduced 24hours after CS (figure 1D). The effects of ivabradine on cardiac function were determined by cardiac ultrasound, which revealed a significant improvement in left ventricular ejection fraction by day 7 after CS in animals treated with ivabradine (figure 1E). This improvement was closely correlated with a decline in the left ventricular necrotic area, as detected by Evans blue/TTC staining (figure 1F).
 and stroke volume (C) after CS. D: cardiac troponin before and 24hours after CS (N=9/group, mean±standard deviation; * P <.05 for ivabradine vs placebo). Left ventricular ejection fraction (N=9/group) (E) and necrotic area (N=5/group) (F) as a percentage of necrotic tissue (pale) vs the area at risk (red) 7 days after CS (mean±standard deviation; * P <.05 for ivabradine vs placebo). CS, cardiogenic shock.")
Ivabradine improves cardiac function in a porcine model of CS. A: schematic representation of the assay. Effects of 0.3mg/kg ivabradine or placebo on heart rate (B) and stroke volume (C) after CS. D: cardiac troponin before and 24hours after CS (N=9/group, mean±standard deviation; * P <.05 for ivabradine vs placebo). Left ventricular ejection fraction (N=9/group) (E) and necrotic area (N=5/group) (F) as a percentage of necrotic tissue (pale) vs the area at risk (red) 7 days after CS (mean±standard deviation; * P <.05 for ivabradine vs placebo). CS, cardiogenic shock.
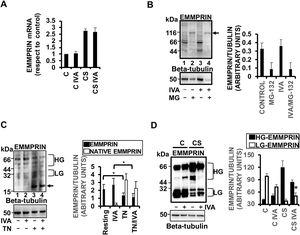
Coronary ischemia/reperfusion induces the expression of several cardiac ECM-degrading enzymes, including MMPs. Administration of 0.3mg/kg ivabradine reduced the left ventricular levels of MMP-9 in the necrotic area by day 7 after CS, as detected by immunohistochemistry and immunoblotting with anti-MMP-9 antibody (figure 2A,B, respectively).
. B: immunoblotting of the 92-kDa pro-MMP-9 from healthy (H), at risk (R), and necrotic (N) sections of hearts subjected to CS (N=5/group, mean±standard deviation; * P <.05 for necrotic ivabradine vs necrotic placebo). CS, cardiogenic shock; MMP-9, matrix metalloproteinase-9.")
Ivabradine reduces the levels of extracellular matrix-degrading enzymes in the heart. A: detection of MMP-9 in the necrotic areas of hearts subjected to CS in response to placebo or ivabradine (N=5/group, mean±standard deviation; * P <.05 for necrotic ivabradine vs necrotic placebo). B: immunoblotting of the 92-kDa pro-MMP-9 from healthy (H), at risk (R), and necrotic (N) sections of hearts subjected to CS (N=5/group, mean±standard deviation; * P <.05 for necrotic ivabradine vs necrotic placebo). CS, cardiogenic shock; MMP-9, matrix metalloproteinase-9.
The role of the MMP inducer EMMPRIN in the expression and activity of MMP-9 has previously been described.3 In our pig model, CS-induced expression of EMMPRIN mRNA was not inhibited by administration of 0.3mg/kg ivabradine (figure 3A). However, ivabradine played a significant posttranslational role, reducing the levels of EMMPRIN and EMMPRIN glycosylation in response to CS. Likewise, ivabradine did not affect the proteolysis of EMMPRIN, as seen by incubation of myocyte H9c2 cell lysates with the proteasome inhibitor MG-132 (figure 3B). Furthermore, incubation of resting H9c2 cells with ivabradine did not inhibit EMMPRIN glycosylation (a key step in EMMPRIN-mediated MMP expression and activity), as seen with treatment of cells with 5μg/mL of the N-linked glycosylation pharmacological inhibitor tunicamycin for 24hours (figure 3C). Similar results were obtained in myocyte HL1B cell cultures (). Conversely, in vivo, and in response to CS, ivabradine significantly reduced the high-glycosylated forms of EMMPRIN (HG-EMMPRIN), resulting in the accumulation of the low-glycosylated forms of EMMPRIN (LG-EMMPRIN), when compared with pigs subjected to CS and treated with placebo, through unknown mechanisms (figure 3D).
. B: immunoblotting of EMMPRIN in H9c2 cardiac cells incubated with MG-132. C: immunoblotting of EMMPRIN in H9c2 cardiac cells incubated with tunicamycin. The arrow indicates the low-molecular-weight 20-kDa native nonglycosylated EMMPRIN. D: expression of EMMPRIN in necrotic areas of hearts subjected to CS and incubated with ivabradine (N=3/group, mean±standard deviation; * P <.05 for HG- vs LG-EMMPRIN with CS + ivabradine). Beta-tubulin was used as a control (B-D).")
Ivabradine reduces the levels of extracellular matrix-degrading enzymes in the heart. A: RT-PCR expression of EMMPRIN mRNA in the hearts of pigs treated with 0.3mg/kg ivabradine. GAPDH was used as a control (N=3/group, mean±standard deviation). B: immunoblotting of EMMPRIN in H9c2 cardiac cells incubated with MG-132. C: immunoblotting of EMMPRIN in H9c2 cardiac cells incubated with tunicamycin. The arrow indicates the low-molecular-weight 20-kDa native nonglycosylated EMMPRIN. D: expression of EMMPRIN in necrotic areas of hearts subjected to CS and incubated with ivabradine (N=3/group, mean±standard deviation; * P <.05 for HG- vs LG-EMMPRIN with CS + ivabradine). Beta-tubulin was used as a control (B-D).
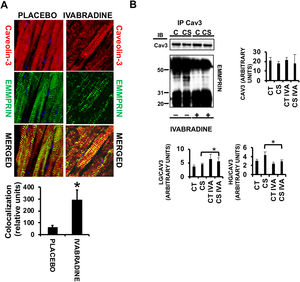
Caveolae are sphingolipid- and cholesterol-enriched vesicles that harbor many signaling pathways playing key roles in cell survival, death, proliferation, and/or migration. In the heart, the principal component in caveolae is caveolin-3, which exerts cardioprotective functions by binding to several proteins. We previously demonstrated that nitric oxide induces cardiac protection by stabilizing the caveolin-3/LG-EMMPRIN complex and thereby preventing HG-EMMPRIN-dependent matrix metalloproteinase-induced cardiac necrosis.14 Here, we detected strong colocalization of caveolin-3 and EMMPRIN in response to 0.3mg/kg ivabradine (figure 4A, ivabradine merged panel, yellow). Protein coimmunoprecipitation assays from the same hearts used in the above experiments revealed an increase in LG-EMMPRIN bound to caveolin-3 (figure 4B) in response to ivabradine, suggesting that ivabradine may reduce CS-induced ECM degradation by preserving the caveolin-3/LG-EMMPRIN complex.
 and EMMPRIN (FITC, green) in heart sections of pigs treated with ivabradine or placebo (colocalization in merged panels=yellow) (N=3/group, mean±standard deviation; * P <.002 for ivabradine vs placebo). B: immunoblotting of caveolin-3 and EMMPRIN in caveolin-3-immunoprecipitated extracts (N=3/group, mean±standard deviation; * P <.05 for CS vs CS IVA).")
Ivabradine prevents disruption of the EMMPRIN/caveolin-3 complex in pigs subjected to cardiogenic shock. A: confocal microscopy detection of caveolin-3 (Cy3, red) and EMMPRIN (FITC, green) in heart sections of pigs treated with ivabradine or placebo (colocalization in merged panels=yellow) (N=3/group, mean±standard deviation; * P <.002 for ivabradine vs placebo). B: immunoblotting of caveolin-3 and EMMPRIN in caveolin-3-immunoprecipitated extracts (N=3/group, mean±standard deviation; * P <.05 for CS vs CS IVA).
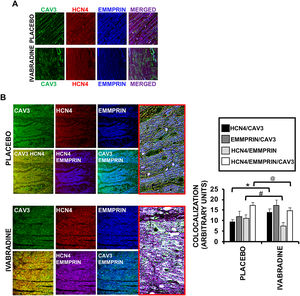
In addition to EMMPRIN, caveolin-3 also binds to HCN4,15 the main target of ivabradine. Confocal microscopic visualization of caveolin-3 in ventricular sections of healthy hearts enabled the detection of a protein complex comprising caveolin-3, EMMPRIN, and HCN4 (figure 5A), although an even more intriguing discovery was the finding that, after CS, the association between caveolin-3/EMMPRIN and caveolin-3/HCN4 was increased in the presence of ivabradine vs placebo (figure 5B). This result suggests that caveolin-3 may be a target of ivabradine in cardioprotection by, on the one hand, preventing degradation of the ECM via increased binding to EMMPRIN and, on the other, stabilizing the complex with HCN4 and reducing the heart rate, as previously seen (figure 1). An interesting discovery is the formation of a HCN4 and EMMPRIN complex and the ability of ivabradine to uncouple the binding of the 2 proteins.
![Ivabradine regulates the binding of caveolin-3, HCN4, and EMMPRIN. A: confocal microscopy detection of caveolin-3 (FITC [green]), HCN4 (TRITC 561 [red]), and EMMPRIN (Alexa Fluor 350 [blue]) in heart sections of healthy pigs (A) or in pigs 7 days after CS (B) and treated with 0.3mg/kg ivabradine or placebo (N=3/group, mean±standard deviation; * P <.05 for HCN4/CAV3 placebo vs ivabradine; #P <.002 for HCN4/EMMPRIN placebo vs ivabradine; @P <.05 for HCN4/EMMPRIN/CAV3 placebo vs ivabradine). CS, cardiogenic shock; FITC, fluorescein isothiocyanate; TRITC, tetramethylrhodamine-isothiocyanate.](https://static.elsevier.es/multimedia/18855857/0000007400000012/v4_202203130557/S1885585720304151/v4_202203130557/en/main.assets/gr5.jpeg?xkr=eyJpdiI6ImJsTFR0VTF2TWxJQmRNL0kvRjQ5dVE9PSIsInZhbHVlIjoia1dtcnJPWUNhTzEvSVZnMXRLRlBBUUxueGZiL21IWm82a1paemFBMHF6WXpsbWRCV3ZOTzNjWjdmOWhxb3hGVzVDemQzYms4Z1UyRUpCcmVtODY4STFMZXlvU2daWkMvL0FLdzFXWjl6aEtzREQxdFpMdm5XM1U4dE5BZjlFSXIwTTZpeVNqazNVSythM0l1aDZDYkFYdlUwaFpNVFc5WndJb3p1U0kzMUJkOXQ3Znk4VVBRam9IaVZwZm5NY3VYTWRpSDZSbTNKcUc4NGUxeWxseG1ZQWQzV0Q5M2YwbHJNL1pYNm1DT3RJUHdDVTdacDBOS0ZDeFc2NmdNSzBlZWlaUUpKd0VjS3I1S081Z080RkxkZU5qVktQMG1qd2d1akVmVHoyaVUrc1E9IiwibWFjIjoiYmRhZmQ1NGMxYzk1ZmU5OGY4YzM1OGQ4NWRlYmZlZTMxYjg4ZWExNWQ1MDNhNWQ5OGU4ZmZjN2QxNTQwNWI0MyIsInRhZyI6IiJ9 "Ivabradine regulates the binding of caveolin-3, HCN4, and EMMPRIN. A: confocal microscopy detection of caveolin-3 (FITC [green]), HCN4 (TRITC 561 [red]), and EMMPRIN (Alexa Fluor 350 [blue]) in heart sections of healthy pigs (A) or in pigs 7 days after CS (B) and treated with 0.3mg/kg ivabradine or placebo (N=3/group, mean±standard deviation; * P <.05 for HCN4/CAV3 placebo vs ivabradine; #P <.002 for HCN4/EMMPRIN placebo vs ivabradine; @P <.05 for HCN4/EMMPRIN/CAV3 placebo vs ivabradine). CS, cardiogenic shock; FITC, fluorescein isothiocyanate; TRITC, tetramethylrhodamine-isothiocyanate.")
Ivabradine regulates the binding of caveolin-3, HCN4, and EMMPRIN. A: confocal microscopy detection of caveolin-3 (FITC [green]), HCN4 (TRITC 561 [red]), and EMMPRIN (Alexa Fluor 350 [blue]) in heart sections of healthy pigs (A) or in pigs 7 days after CS (B) and treated with 0.3mg/kg ivabradine or placebo (N=3/group, mean±standard deviation; * P <.05 for HCN4/CAV3 placebo vs ivabradine; #P <.002 for HCN4/EMMPRIN placebo vs ivabradine; @P <.05 for HCN4/EMMPRIN/CAV3 placebo vs ivabradine). CS, cardiogenic shock; FITC, fluorescein isothiocyanate; TRITC, tetramethylrhodamine-isothiocyanate.
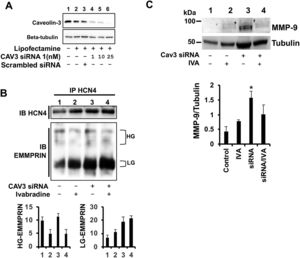
To test whether caveolin-3 can serve as a docking point between HCN4 and EMMPRIN, we silenced the expression of caveolin-3 with caveolin-3-specific siRNAs in H9c2 cardiac cells. With 10 nM siRNA, caveolin-3 expression was reduced by more than 75% vs nontransfected cells or cells transfected with scrambled siRNA (figure 6A). HCN4 bound to EMMPRIN in both caveolin-3-expressing and -silenced cells, suggesting the existence of a direct interaction (figure 6B). On the other hand, incubation with ivabradine yielded a significant uncoupling of HG-EMMPRIN bound to HCN4, independent of caveolin-3 expression (figure 6B). Likewise, the increased expression of MMP-9 in caveolin-3-silenced cells was downregulated by ivabradine (figure 6C), indicating that ivabradine may target caveolin-3 to prevent ECM degradation.
 or 1, 10, and 25nM caveolin-3 siRNA1. Beta-tubulin was used as a negative control. B, upper panel: immunoblotting of HCN4 from extracts immunoprecipitated with HCN4 from H9c2 cells silenced with 10nM caveolin-3 siRNA1; lower panel: immunoblotting of EMMPRIN from the same extracts (N=3). C: immunoblotting of MMP-9 in H9c2-silenced cells incubated with 0.3mg/kg ivabradine (N=3, mean±standard deviation; P <.001 for siRNA vs siRNA + ivabradine). MMP-9, matrix metalloproteinase-9; siRNA, small interfering RNA.")
Lack of caveolin-3 impairs formation of the EMMPRIN/HCN4 complex. A: immunoblotting of caveolin-3 from H9c2 cells transfected with Lipofectamine (no siRNA) or 1, 10, and 25nM caveolin-3 siRNA1. Beta-tubulin was used as a negative control. B, upper panel: immunoblotting of HCN4 from extracts immunoprecipitated with HCN4 from H9c2 cells silenced with 10nM caveolin-3 siRNA1; lower panel: immunoblotting of EMMPRIN from the same extracts (N=3). C: immunoblotting of MMP-9 in H9c2-silenced cells incubated with 0.3mg/kg ivabradine (N=3, mean±standard deviation; P <.001 for siRNA vs siRNA + ivabradine). MMP-9, matrix metalloproteinase-9; siRNA, small interfering RNA.
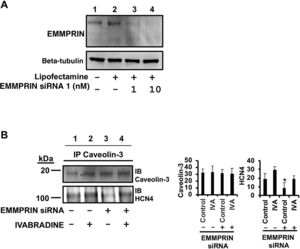
In the same way, we also used EMMPRIN-specific siRNAs to investigate the role of EMMPRIN in the complex between caveolin-3 and HCN4 (figure 7A). Inhibition of EMMPRIN reduced the levels of the HCN4/caveolin-3 complex, as determined by immunoblot detection of HCN4 in caveolin-3-immunoprecipitated cell lysates of H9c2 cells (figure 7B, line 3). This result indicates that EMMPRIN is required to maintain the HCN4/caveolin-3 complex and that their interaction is partly restored by incubation with ivabradine (figure 7B, lane 4), suggesting that EMMPRIN-independent binding of caveolin-3 and HCN4 occurs in response to ivabradine.
. siRNA, small interfering RNA.")
Lack of EMMPRIN impairs formation of the caveolin-3/HCN4 complex. A: gene silencing of EMMPRIN with EMMPRIN-specific siRNA. Beta-tubulin was used as a loading control. B: immunoblotting of HCN4 from extracts immunoprecipitated with anti-caveolin-3 from H9c2 cells silenced with 10nM EMMPRIN siRNA1 (N=3, mean±standard deviation; P <.004 for HCN4 siRNA vs siRNA + ivabradine). siRNA, small interfering RNA.
In the current work, we found that ivabradine induces cardiac protection beyond the I(f) current. Under CS conditions, ivabradine administration may help to prevent cardiac necrosis by reducing the levels of the ECM-degrading enzyme MMP-9, as well as EMMPRIN levels and EMMPRIN glycosylation (required for MMP-induced degradation of ECM), through stabilization of the caveolin-3/LG-EMMPRIN complex and the binding of HCN4, the main target of ivabradine, to LG-EMMPRIN. Ivabradine also stabilized the HCN4/caveolin-3 complex, suggesting a bradycardic effect beyond the I(f) current. Taken together, we propose a new molecular mechanism in which administration of ivabradine promotes cardioprotection after CS.
Ischemia/reperfusion injury induces ECM degradation and cardiac necrosis through the expression of MMPs in cardiac cells,16–18 at least partly via activation of EMMPRIN.13 Targeting of EMMPRIN not only reduces the ventricular expression of MMP-9, but also successfully prevents necrosis progression, thereby improving the left ventricular ejection fraction after reperfusion.19 High-glycosylated forms of EMMPRIN (HG-EMMPRIN) induce MMP-9 expression,13 whereas LG-EMMPRIN remains bound to caveolin-3 in cardiac myocytes, preventing downstream MMP expression, as we previously reported.14 Here, we found that, under CS conditions, administration of ivabradine stabilized the EMMPRIN/caveolin-3 complex, suggesting that, in addition to the I(f) current, and given that beta-adrenergic stimulation triggers the expression of EMMPRIN in cardiac myocytes,20,21 inhibition of EMMPRIN may also exert cardioprotective effects by inhibiting beta-adrenergic heart rate stimulation in our CS model.
The effect of ivabradine on MMP expression was previously reported in diabetic mice.22 In that model, ivabradine reduced the levels of MMP-2 and improved cardiac function by still unknown mechanisms. In agreement with those findings, our study has discovered a cardioprotective mechanism elicited by ivabradine beyond the I(f) current that involves an in vivo reduction in the total amount and glycosylation of EMMPRIN, required to induce EMMPRIN-mediated MMP activity.
Our data show that EMMPRIN is induced under hypoxic conditions in cardiac cells, as others found in human embryonic kidney cells,23 microglia,24 and different types of cancer cell cultures.25,26 Hypoxia is a powerful stimulus of necrosis and apoptosis in several tissues. Cardiac cells respond to hypoxia by triggering several signaling pathways, including the stabilization of specific proteins via inhibition of ubiquitination-mediated degradation, as recently reported for caveolin-3, which clarifies the role of caveolin-3 in cardioprotection against ischemic injury.27 We show that, in response to CS, ivabradine stabilizes caveolin-3 and thereby preserves the caveolin-3/LG-EMMPRIN complex, preventing MMP-dependent ECM degradation. Therefore, we propose that ivabradine helps to stabilize caveolin-3 under ischemic conditions.
The use of ivabradine is based on its ability to inhibit the HCN4-dependent I(f) current in cardiomyocytes by thus far unknown mechanisms. Caveolin-3 binds to HCN4 in cardiac caveolae, which enables it to regulate pacemaker activity.15 Dysregulation of the caveolin-3/HCN4 complex has been linked to several types of arrhythmogenic behavior, as recently reported through the expression of the caveolin-3 variant T78M28 or in certain phenotypes of long QT syndrome, through acceleration of HCN4 activation kinetics.29 In agreement with the literature, our data indicate that ivabradine induces cardiac protection by preserving I(f) conductance through stabilization of the caveolin-3/HCN4 complex.
Thus far, no studies have addressed the involvement of EMMPRIN in the caveolin-3/HCN4 complex. Interestingly, our data suggest a direct interaction between HCN4 and EMMPRIN, with ivabradine destabilizing the complex. The interplay between HCN4 and EMMPRIN remains unknown, and further investigation is warranted to determine whether the binding is merely circumstantial or whether it represents a new target in necrosis. Alternatively, we cannot exclude the possibility that HCN4 and EMMPRIN bind to common partners and not via a direct interaction. Such is the case for beta-adrenergic receptors, which bind to HCN430 and EMMPRIN.31 In this pathway, beta-adrenergic stimulation activates HCN4 and is related to the inflammatory actions of EMMPRIN.
LimitationsOne of the study limitations is the use of female pigs because females and males have different cardioprotective physiologies,32 which may compromise the level of the heart injury and heart failure related to the ischemic event. The use of rat H9c2 cell cultures is also a limitation. Isolation of pig cardiac myocytes would be the best experimental approach for testing the specific parameters addressed in this work. However, the extremely difficult isolation of living porcine cardiac myocytes led us to use H9c2 and HL1B cells to assess hypoxia and reoxygenation.
CONCLUSIONSIvabradine induces cardiac protection beyond direct inhibition of the I(f) current. During CS, ivabradine stabilizes the HCN4/caveolin-3 complex and prevents cardiac necrosis by preserving LG-EMMPRIN. Ivabradine also stabilizes the caveolin-3/HCN4 complex, already reported as a mechanism to regulate the I(f) current. Further studies will explore the molecular role of ivabradine in EMMPRIN and HCN4 dimerization as a new therapeutic tool.
FUNDINGThis work was supported by the Universidad Francisco de Vitoria (grant numbers 2017/18), Fundación BBVA (2017), and Proyectos de +D+I, from the program Investigación orientada a los retos de la sociedad (MINECO/AEI/FEDER/EU [SAF2017-87342-R] to CZ).
CONFLICTS OF INTERESTNone.
- -
Ivabradine, a selective inhibitor of the pacemaker I(f) current in sinoatrial cells, inhibits HCN1 and HCN4 channels, reduces heart rate, and preserves blood pressure and left ventricular function and is thus recommended in the treatment of chronic heart failure and left ventricular ejection fraction <35%; however, its effectiveness in acute heart failure is unclear.
- -
Cardiogenic shock is a frequent cause of death after acute myocardial infarction.
- -
Vasoactive drugs may contribute to myocardial damage through extensive tachycardia and increased oxidative stress and oxygen demand.
- -
We previously found that ivabradine improves catecholamine-induced sinus tachycardia under cardiogenic shock conditions, improving hemodynamic parameters by unknown mechanisms.
- -
Ivabradine inhibits the HCN-dependent I(f) current by mechanisms yet to be addressed; caveolin-3 binds to HCN4 in cardiomyocytes to regulate pacemaker activity.
- -
Our data suggest that ivabradine exerts cardioprotective effects by preserving I(f) conductance through stabilization of the caveolin-3/HCN4 complex.
- -
Caveolin-3 induces cardiac protection by forming a complex with LG-EMMPRIN, providing a new molecular mechanism for cardiac protection involving prevention of ECM degradation in the heart.
- -
Further research is required to investigate findings related to the HCN4/EMMPRIN complex during CS.
Supplementary data associated with this article can be found in the online version available at https://doi.org/10.1016/j.rec.2020.09.012