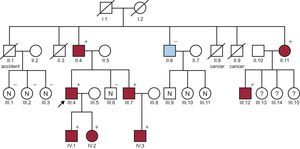
We present a family with 8 members from 3 generations with Marfan syndrome (MFS), in whom a complete familial genetic study confirmed the pathogenicity of a variant previously thought to be “synonymous”, identified on the fibrillin 1 gene (FBN1).
The proband is a 50-year-old Caucasian male who underwent emergency surgery for a type A aortic dissection. His 79-year-old father underwent surgery at age 56 years for dilatation of the ascending aorta with replacement of the aorta and aortic valve (Bentall procedure) and was operated on again at age 75 years for dilatation of the descending aorta. The proband had been diagnosed with ectopia lentis prior to his aortic dissection. In view of this and his father's history, MFS was suspected. In 2008, he underwent a genetic study of the FBN1 gene in another center, using Sanger sequencing and multiplex ligation-dependent probe amplification (MLPA), but this was reported as negative. His 2 children were under follow-up as suspected MFS in a third center.
The familial study was centralized in our hospital. When the family tree was drawn (Figure), it identified other members with aortic dilatation or variable expression of features of MFS (Table).1 The younger brother, who also had ectopia lentis, recently underwent surgery for a descending aortic aneurysm, and received a Bentall valved tube. This patient declined the aortic valve-sparing surgery offered (David procedure) due to the good outcomes in his father and brother, who had the Bentall procedure.

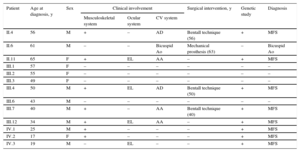
Clinical Information on the Family Members Studied
| Patient | Age at diagnosis, y | Sex | Clinical involvement | Surgical intervention, y | Genetic study | Diagnosis | ||
|---|---|---|---|---|---|---|---|---|
| Musculoskeletal system | Ocular system | CV system | ||||||
| II.4 | 56 | M | + | – | AD | Bentall technique (56) | + | MFS |
| II.6 | 61 | M | – | – | Bicuspid Ao | Mechanical prosthesis (63) | – | Bicuspid Ao |
| II.11 | 65 | F | + | EL | AA | – | + | MFS |
| III.1 | 57 | F | – | – | – | – | – | – |
| III.2 | 55 | F | – | – | – | – | – | – |
| III.3 | 49 | F | – | – | – | – | – | – |
| III.4 | 50 | M | + | EL | AD | Bentall technique (50) | + | MFS |
| III.6 | 43 | M | – | – | – | – | – | – |
| III.7 | 40 | M | + | – | AA | Bentall technique (40) | + | MFS |
| III.12 | 34 | M | + | EL | AA | – | + | MFS |
| IV.1 | 25 | M | + | – | – | – | + | MFS |
| IV.2 | 17 | F | + | – | – | – | + | MFS |
| IV.3 | 19 | M | – | EL | – | – | + | MFS |
–, negative; +, positive; AA, aortic aneurysm; AD, aortic dissection; Ao, aorta; CV, cardiovascular; EL, ectopia lentis; F, female; M, male; MFS, Marfan syndrome.
Musculoskeletal system + if any of the following features were present: arachnodactyly, scoliosis, chest deformity, striae, acetabular protrusion, plat foot or foot deformity.
Given the obvious familial disease, the negative results of the previous genetic study were striking. In MFS, the percentage of patients with a positive genetic study has been reported as close to 90%.2 Another genetic study was performed, this time using next generation sequencing with a panel that included 30 genes involved in aortic disease. This study identified only 1 “synonymous” variant (meaning that the change in nucleotide does not produce a change in the amino acid) in the FBN1 gene (c.6354C>T, p.Ile2118=). The fact that this does not produce a change in the amino acid could indicate that this variant is nonpathogenic or of borderline pathogenicity. It is located in a coding region of the gene (at position 40 of exon 52, isoform NM_000138.4), a position that does not appear to affect any of the sites involved in the RNA cutting and splicing process. However, an RNA study, carried out by Liu et al.3 in 1977, showed that this change induced skipping of exon 52 (absence of this exon in the RNA transcript). This may create a new reading frame, leading to a premature stop codon, in turn leading to a truncated protein. Alternatively, the FBN1 protein may not be expressed to the truncated protein because the cellular machinery breaks down the aberrant transcript. The fact that splicing is affected and that functional effects have been demonstrated is sufficient to demonstrate that the mutation is pathogenic.4 Furthermore, this variant does not appear in databases that include information from the population used as a control. To definitively confirm its pathogenicity, however, cosegregation would need to be demonstrated in 1 of the families studied. Although 8 reports have been published of different families with suspected or clinically diagnosed MFS, these reports do not provide data from a familial study, only from the probands.5,6
In the family we present here, the cosegregation of this mutation with the disease has been demonstrated for the first time. All of the members studied meet the revised Ghent nosology4 (Table). There was only 1 patient (II.6) in whom the variant was not identified, a paternal uncle of the proband. It was confirmed that he had an unrelated disease (bicuspid aorta) and had undergone surgery for aortic stenosis. Three daughters of an affected individual (III.13,14,15) could not be studied due to social reasons.
In conclusion, we have been able to establish cosegregation of the p.Ile2118= variant on the FBN1 gene for the first time through a complete clinical and genetic familial study, thus confirming its pathogenicity. We do not know if the negative genetic study performed years before failed to identify this variant (Sanger false negative) or if it was considered nonpathogenic.
It is important to have MFS reference centers or familial heart disease units that allow the performance of complete familial studies.2 Performing a familial study and the correct interpretation of the variants identified in genetic studies allows a more accurate prognosis to be established, and helps ensure appropriate clinical management.
FUNDINGThis study was partly funded by the Cardiovascular Research Network, Red de Investigación Cardiovascular (RIC; RD12/0042/0069).
CONFLICTS OF INTERESTJ.P. Trujillo-Quintero belongs to the clinical department of Health in Code.