


Cardiac channelopathies are genetic alterations that can cause sudden death. Long QT syndrome and Brugada syndrome are 2 such conditions. Both are diagnosed according to previously published criteria. Our objective was to determine the sensitivity of these criteria in a consecutive series of patients carrying the mutations that cause them.
MethodsWe enrolled 15 families and 31 causal mutation carriers with a high pathogenic probability of having long QT syndrome and Brugada syndrome. We conducted clinical and electrocardiographic studies to analyze the extent to which these patients fulfilled the diagnostic criteria. Statistical analysis was with SPSS 17.0.
ResultsSome 48.3% of the subjects met the criteria indicating a high probability of long QT syndrome or Brugada syndrome. Among those with the mutation for long QT syndrome, only 10 out of 21 had a Schwartz index score≥4. Both the median Schwartz score and the cQT interval were lower in relatives than in probands. Of those with the mutation for Brugada syndrome, 60% failed to meet current diagnostic criteria, which were more frequently fulfilled in relatives. Pharmacological tests with epinephrine and flecainide helped establish the diagnosis in 2 mutation carriers with negative phenotype.
ConclusionsCurrent diagnostic criteria for long QT syndrome and Brugada syndrome had low sensitivity in our sample of genetic carriers. Genetic tests supported by pharmacological tests can increase diagnostic sensitivity, especially in asymptomatic relatives.
Keywords
.
INTRODUCTIONSudden death in persons with a structurally normal heart (arrhythmic sudden death syndrome) is usually arrhythmic in origin and is primarily caused by channelopathies or alterations of the cardiac ion channels.1 The most prevalent of these are long QT syndrome (LQTS), Brugada syndrome (BS), catecholaminergic polymorphic ventricular tachycardia and idiopathic ventricular fibrillation (IVF).2 Characteristics common to channelopathies are a defined genetic origin, a structurally normal heart, predisposition to ventricular arrhythmias and sudden death, and usually incomplete clinical penetrance.3 Exhaustive cardiological evaluation with surface electrocardiogram (ECG) and pharmacological tests with flecainide and epinephrine successfully identify the underlying etiology of an arrhythmic episode in a high proportion of cases.4–6 Cases of sudden death without a final diagnosis despite the performance of conventional tests are classified as IVF.
Diagnosis of channelopathies—specifically diagnosis of LQTS and BS—is performed according to existing criteria that remain current at the time of writing (Table 1). The Schwartz criteria, published in 1985 and modified in 1993,7 consider diagnosis of LQTS as probable when the score is≥4 and take into account electrocardiographic, clinical and family history data (Table 1A). For BS, the criteria applied since 20058 require the presence of Brugada type 1 pattern in at least 2 precordial leads of the baseline surface ECG or after intravenous infusion of a sodium blocker (Table 1B).
Long QT Syndrome and Brugada Diagnostic Criteria.
| Points | |
| A) LQTS diagnostic criteria | |
| ECG findings | |
| cQT calculated using Bazzet's formula>480 ms | 3 |
| 460-40 ms | 2 |
| 450 ms, men | 1 |
| Torsade de pointes | 2 |
| T wave alternans | 1 |
| Bifid T wave in 3 leads | 1 |
| Baseline heart rate<2 centiles of the level corresponding to age | 0.5 |
| Clinical history | |
| Syncope with stress | 2 |
| Syncope without stress | 1 |
| Congenital deafness | 0.5 |
| Family history | |
| First-degree relatives with definitive LQTS according to Schwartz criteria | 1 |
| First-degree relatives with sudden death at less than 30 years | 0.5 |
| B) BS diagnostic criteria | |
| ECG criteria (necessary) | One of the following must be present |
| Brugada type 1 pattern in 2 or more leads (V1-V3) in presence or absence of an Na channel blocker | Documented VF |
| Documented polymorphic VT | |
| Family history of sudden death at less than 40 years | |
| Type 1 ECG pattern in close relatives | |
| VT induced in EPS | |
| Syncope | |
| Nocturnal agonal respiration |
BS, Brugada syndrome; ECG, electrocardiogram; EPS, electrophysiologic study; LQTS, long QT syndrome; VF, ventricular fibrillation; VT, ventricular tachycardia.
LQTS≥4 points: high probability; LQTS 2-3 points: intermediate probability; LQTS≤1 point: low probability.
Neither of these criteria takes account of genetic study data. Recent research points to the importance of the genetic study combined with the phenotype-oriented pharmacological test in diagnosing cases that are dubious because of the incomplete clinical penetrance of these alterations, especially in asymptomatic relatives.9–13 The objective of our study was to analyze the validity of the diagnostic criteria in patients diagnosed as having a high probability of a channelopathy on the basis of genetic test results and to study the value of these diagnostic criteria in the proband and relatives, supported by the pharmacological test in genetic mutation-carriers with negative phenotype.
METHODSWe studied index cases suspected of having a channelopathy because they had 1 or more of the following characteristics: a history of polymorphic ventricular arrhythmias of uncertain etiology or cardiogenic syncope, an ECG suggestive of LQTS or BS, or they were a close relative of someone who died of probable arrhythmic sudden death or arrhythmic sudden death syndrome (autopsy with no findings of structural abnormalities and context of death indicating sudden cardiac death). All subjects underwent a genetic study to sequence the genes most prevalent in LQTS and BS. We only included those probands who were causal mutation carriers. Close relatives underwent ECG and genetic testing to identify or exclude the presence of the same mutation as that found in the proband. We enrolled those relatives who were mutation carriers and excluded noncarriers and those with a reversible secondary cause of prolonged cQT interval (Fig. 1).

Patient enrollment algorithm. We excluded index cases and relatives with negative genotype. Note that the mutational prevalence was similar to that of previous studies, with 70.5% for long QT syndrome and 30% for Brugada syndrome. BS, Brugada syndrome; ECG, electrocardiogram; LQTS, long QT syndrome.
The mutation found had to be associated with a high probability of LQTS or BS either because it had been previously described or because it showed molecular characteristics—such as the location in the protein, resulting amino acid change or described effects of mutations in zones near the same gene—indicating that it had a high probability of being pathogenic.14
All subjects underwent 25 and 50mm/s ECGs, the cQT interval being measured with the Bazzet formula15 and signs of Brugada type 1 pattern (>1mm ST segment elevation in at least 2 precordial leads with typical morphology) being sought. All subjects underwent transthoracic echocardiography to rule out structural heart disease. Those with no phenotype in the ECG underwent pharmacological tests with epinephrine and/or flecainide13,16 depending on the genotype and/or suspected epidemiological factors (for BS, sudden death in a relative at rest; for LQTS, situations involving a high level of physical effort, physical or emotional stress leading to syncope, syncope or ventricular arrhythmias in response to auditory stimuli or in relation to bathing for different LQTS subtypes, etc.). We calculated the Schwartz index in carriers of the genetic mutation causing LQTS and classified them in 3 groups according to whether or not they met the diagnostic criteria for BS.
Statistical AnalysisThe statistical analysis was performed with SPSS 17.0 (Chicago, United States). Descriptive variables were used to study the proportion of subjects who met the diagnostic criteria for each illness (measures of central tendency, such as mean and median, and of proportion, such as percentages). We later conducted a statistical comparison using nonparametric tests (the Mann-Whitney U test and the Fisher exact text) between probands and close relatives to find differences in the variables of phenotype expression.
This study was approved by the hospital ethics committee and all patients gave signed informed consent for the genetic sequencing study.
RESULTSInitially, we evaluated 27 index cases with suspected channelopathy (Fig. 1); 17 with suspected LQTS and 10 with suspected BS. We excluded 5 patients with LQTS and no genetic confirmation of the diagnosis (70.5% mutational prevalence) and 7 index cases with a clinical diagnosis of BS showing no mutations in SCN5A (30% mutational prevalence). Of the 12 index cases with LQTS, consultation had been motivated by syncope in 6 and cardiac arrest by ventricular fibrillation in 4; 2 were diagnosed after they underwent investigation due to the sudden cardiac death of a close relative. Of the 3 probands with BS, 2 had had a cardiac arrest and 1 was asymptomatic.
The mutations found in patients with LQTS were principally located in the KCNH2 gene; we found 1 mutation in KCNQ1; in 2 subjects the gene affected was SCN5A. These mutations have already been described by our group elsewhere.10,17 In BS, the 3 mutations found had not previously been described and are shown in Table 2. Of all the positive cases, 2 had undergone genetic study for IVF, with normal results of ECG and pharmacological tests.
Some 20% of the mutations found had previously been described as associated with LQTS; in another 2 patients, our group studied the electrophysiological properties of the mutated channel in a cellular model. We based the pathogenicity of the other mutations on the aforementioned data.14,17
After including first- and second-degree relatives who showed the same mutation considered highly likely to be pathogenic, we finally included 31 mutation carriers (21 carriers of LQTS, 12 probands and 9 relatives; and 10 carriers of BS, 3 probands and 7 relatives).
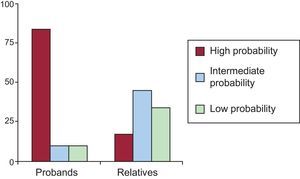
Fifteen of the 31 met the diagnostic criteria for the channelopathy in question (Table 1), representing 48.3% of the total; 11 of 15 probands (73.3%) and 4 of 16 relatives (25%). The genetic study identified the underlying channelopathy in 4 probands with indeterminate phenotype and detected silent carrier status in 12 relatives with no phenotypic expression. Below, and in Tables 3 and 4, we present our results by channelopathy type together with our comparison of phenotypic expression in probands and relatives.
Phenotype Evaluation in Long QT Syndrome Mutation Carriers.
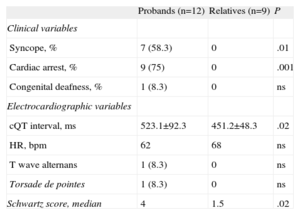
| Probands (n=12) | Relatives (n=9) | P | |
| Clinical variables | |||
| Syncope, % | 7 (58.3) | 0 | .01 |
| Cardiac arrest, % | 9 (75) | 0 | .001 |
| Congenital deafness, % | 1 (8.3) | 0 | ns |
| Electrocardiographic variables | |||
| cQT interval, ms | 523.1±92.3 | 451.2±48.3 | .02 |
| HR, bpm | 62 | 68 | ns |
| T wave alternans | 1 (8.3) | 0 | ns |
| Torsade de pointes | 1 (8.3) | 0 | ns |
| Schwartz score, median | 4 | 1.5 | .02 |
HR, heart rate; ns, not significant.
The data are expressed as mean±standard deviation or no. (%).
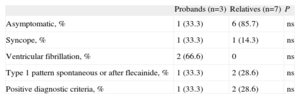
Phenotype Evaluation in Brugada Syndrome Mutation Carriers.
| Probands (n=3) | Relatives (n=7) | P | |
| Asymptomatic, % | 1 (33.3) | 6 (85.7) | ns |
| Syncope, % | 1 (33.3) | 1 (14.3) | ns |
| Ventricular fibrillation, % | 2 (66.6) | 0 | ns |
| Type 1 pattern spontaneous or after flecainide, % | 1 (33.3) | 2 (28.6) | ns |
| Positive diagnostic criteria, % | 1 (33.3) | 2 (28.6) | ns |
ns, not significant.
Of 21 subjects with the LQTS genotype, only 12 (10 probands and 2 relatives) met the Schwartz diagnostic criteria with high probability of LQTS (57.1%). Two had an intermediate probability of LQTS (9.5%) and 7 (33.3%) a low probability. Figure 2 presents the differences in Schwartz scores between probands and relatives. The mean age was 24.1 (17) years; 41.7% were men. We found no differences in phenotypic expression as a function of age or sex.

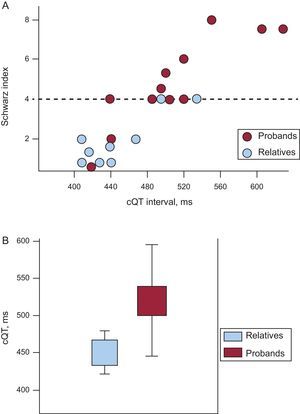
As Figure 3A shows, the high proportion of subjects who did not meet LQTS criteria indicated cQT interval values that were generally normal or at the upper limit of normality, giving them final Schwartz scores of <4. Spearman's rho correlation coefficient for both variables was 0.89, indicating a high correlation between the parameters. Subgroup analysis (Fig. 3A) showed that the proportion of subjects presenting Schwartz scores<4 was significantly greater among close relatives, who were usually asymptomatic, than among index cases. The median Schwartz score for probands was 4 vs 1.5 (P=.02) among relatives. Most (7 out of 9) asymptomatic relatives had a Schwartz score<4.
 and relatives (blue); note the good correlation between cQT interval values and the Schwartz index; the dotted line represents a score of 4 on the index above which diagnosis of long QT syndrome is probable; note the high number of relatives who score<4 and the 2 index cases, both with a history of ventricular fibrillation, who score<2. B: comparison of cQT interval length of probands and relatives.")
A: graph of cQT interval distribution in probands (red) and relatives (blue); note the good correlation between cQT interval values and the Schwartz index; the dotted line represents a score of 4 on the index above which diagnosis of long QT syndrome is probable; note the high number of relatives who score<4 and the 2 index cases, both with a history of ventricular fibrillation, who score<2. B: comparison of cQT interval length of probands and relatives.
Among index cases, the mean cQT interval was 523.1 (92.3)ms vs 451.2 (48.3)ms among relatives (P<.05) (Fig. 3B). Not all probands had a prolonged cQT interval or a Schwartz score≥4. Indeed, 2 index cases who had presented episodes of ventricular fibrillation showed normal baseline ECGs and Schwartz scores indicating a low probability of LQTS. In these 2, the adrenalin test was negative and the diagnosis of LQTS was given by the genetic test.
Brugada SyndromeThe mean age was 43.5 (12.5) years; 72.1% were men. We found no differences in phenotypic expression as a function of age or sex.
Of the 10 subjects who carried a mutation causing BS in SCN5A, only 3 met the current diagnostic criteria for this channelopathy. The percentage was greater among probands than among close relatives (Table 4). Of the 3 index cases, 2 did not meet the diagnostic criteria as they did not even show Brugada pattern after intravenous flecainide administration; 1 of them had presented with ventricular fibrillation (Family 2 in Table 2). In this patient, diagnosis was given by the genetic study and the pharmacological test in 2 close relatives who did meet diagnostic criteria as they showed the Brugada type 1 pattern after flecainide administration (Fig. 4). In the other, the ECG showed the type 1 pattern in 1 lead only and therefore the patient did not meet the diagnostic criteria either.
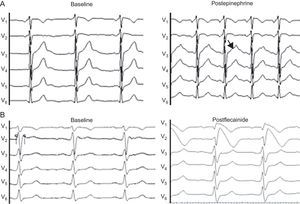
. A: corresponds to a family member who is a carrier of the mutation in KCNH2, and the brother of a man with sudden death and autopsy without pathologic findings; note the notched T wave in the ascending branch after adrenalin infusion, a characteristic sign of type 2 long QT syndrome.16 B: flecainide test in a close relative of a woman with ventricular fibrillation and normal surface electrocardiogram; the baseline electrocardiogram already indicates Brugada syndrome.")
Pharmacological test in dubious cases (electrocardiogram at 50 mm/s). A: corresponds to a family member who is a carrier of the mutation in KCNH2, and the brother of a man with sudden death and autopsy without pathologic findings; note the notched T wave in the ascending branch after adrenalin infusion, a characteristic sign of type 2 long QT syndrome.16 B: flecainide test in a close relative of a woman with ventricular fibrillation and normal surface electrocardiogram; the baseline electrocardiogram already indicates Brugada syndrome.
We compared the baseline ECGs of 6 carriers of a genetic mutation in SCN5A who did not show the Brugada type 1 pattern at baseline or after flecainide administration with those of close relatives who did not have the mutation and found that PR (186.2 [34] vs 153.6 [26]ms) and QRS (109.1 [23] vs 88.4 [13]ms) intervals were significantly prolonged in those who carried the muatation in SCN5A compared with the control subjects (P<.05). We found no ECG showing Brugada type 2 pattern, and the cQT interval was similar in both groups.
Pharmacological TestsIn probands and/or relatives with normal ECGs or who did not meet the diagnostic criteria, the pharmacological test with epinephrine and/or flecainide was performed as a result of clinical suspicion. We conducted 3 epinephrine tests in carriers of mutations in LQTS genes (2 in KCNH2 and 1 in SCN5A) and 8 flecainide tests in carriers in SCN5A. Of these, the epinephrine test helped to confirm the diagnosis in 1 family with a mutation in KCNH2; all members of this family were asymptomatic and were related to a patient with sudden death at 25 years (Fig. 4A). In BS, the flecainide test revealed type 1 pattern in 2 close relatives of the proband with IVF and 1 mutation in SCN5A; the diagnosis was confirmed in all 3 relatives (Fig. 4B). In the remaining cases, the results of the pharmacological test were negative.
DISCUSSIONDiagnosing alterations of the cardiac ion channels is not always simple. Consequently, years ago, diagnostic criteria were established which remain current today (Table 1).7,8 The finding that numerous patients with a confirmed genetic diagnosis of LQTS and BS did not meet the diagnostic criteria is not recent, although no specific study has verified the sensitivity of these criteria in a genotyped sample of causal mutation carriers.3,18 In BS, a recent study indicated the need for a review to include those cases showing the type 1 pattern in a single lead, which would increase the diagnostic sensitivity in our series as 1 genotyped proband showed the pattern in a single lead.19
We present a series of LQTS and BS causal mutation carriers in whom we studied each diagnostic criterion. Our main finding was that fewer than half of the carriers would be identified if we did not have access to the genetic study supporting the pharmacological tests. This finding was most marked in asymptomatic relatives since >70% did not fulfill the diagnostic criteria, confirming that penetrance—ie, phenotypic expression in carriers of a mutation that is probably causal—is low. However, not all of these asymptomatic relatives can be considered patients simply because they carry a genetic mutation since some carriers never express the phenotype and others do so only in situations that alter repolarization, such as taking drugs or ion alterations (silent carriers). It is difficult to identify the genetic carriers who will express the phenotype and those who will not and pharmacological tests can prove to be essential, as our study shows. The dysfunction generated in the ion channel can be of a different level in each case and, moreover, its functional repercussion depends on whether it can be partly or wholly compensated by other ion currents. The finding that relatives who are genetic carriers of a mutation in SCN5A, despite having a negative phenotype for BS, show prolonged PR and QRS intervals compared with relatives who are noncarriers, indicates a certain degree of sodium channel dysfunction.
Having a silent channelopathy or low penetrance does not rule out the risk of ventricular arrhythmias or sudden death.20–22 Priori et al.3 reported that in some families with a history of sudden death clinical penetrance can be extremely low for LQTS. Similarly, the CASPER registry of IVF showed that the combined use of pharmacological and genetic tests revealed a subclinical channelopathy in 24 of 63 patients with normal baseline ECGs.9 In our study, 2 probands with LQTS and 2 with BS did not meet the diagnostic criteria; 3 of them had presented as IVF. This finding reinforces the diagnostic value of genetic testing in symptomatic probands in whom conventional tests fail to identify the causal channelopathy. Although a cQT interval>500ms and spontaneous Brugada type 1 pattern appear to increase the risk of sudden death,23,24 the absence of these alterations in the surface ECG reduces the risk of arrhythmias but does not completely rule it out. Hence the importance of detecting cases in asymptomatic relatives to establish preventative measures, such as avoiding drugs and situations that could potentially induce arrhythmias, and considering beta blocker administration in LQTS.
Identifying a mutation in a subject with a suspected channelopathy should be interpreted appropriately, especially all in asymptomatic relatives. Our study suggests that genetic testing can be decisive in diagnosing a hidden channelopathy in probands. However, a genetic finding without clear repercussions or family cosegregation with the disease can lead to error.14,25 A team of experts in cardiovascular genetics should therefore evaluate and interpret the genetic mutations found. All mutations present in our series showed a very high probability of causing LQTS or BS. We base this argument on the position of the affected amino acid in the protein, the resulting type of amino acid change, and the expected alteration of the protein structure.14 In some cases, the mutation had been previously described in the literature, sometimes with an in vitro study of the electrophysiological properties of the channel, and the conclusion about its possible pathogenic effects in these cases was more obvious.17,26,27
Current diagnostic criteria do not take account of genetic findings when establishing the probability of having a particular channelopathy. In our series of genetic carriers, the sensitivity of current criteria was <50%, even though, as previously mentioned, not all carriers are likely to have the phenotype in the future. Although sodium channel blocker response is included in the diagnostic criteria for BS, in LQTS, the epinephrine test is not. The latter, validated in protocols by Shimizu et al. and the Mayo Clinic,16,28,29 increases the diagnostic precision of dubious or difficult-to-interpret genetic findings, especially in LQTS type 1. These authors suggest that the epinephrine test contributes a high negative predictive value (≤96%) to rule out any chances that the genetic finding may be coincidental and to clearly identify a phenotype hidden in the baseline ECG. In our series (Fig. 2A), we found carriers of mutations in SCN5A and KCNH2 with normal cQT interval. Using the epinephrine test in 1 of these families—even though the mutation was not in KCNQ1, but in KCNH2—led to successful diagnosis in 3 affected relatives (Fig. 4B). As our final results show, pharmacological testing with epinephrine or flecainide should be considered in negative phenotype probands and in relatives. Occasionally, as in the case of BS shown in Figure 4A, the index case showed the negative phenotype and did not fulfill the diagnostic criteria and therefore a diagnosis was not obtained. Studying relatives through the flecainide test enabled us to diagnose BS in the proband who shared the mutation with the silent carriers. However, in light of our results, pharmacological test findings should be interpreted with caution: the absence of an alteration in response to epinephrine or flecainide in a subject who is a genetic carrier does not imply the absence of disease since the epinephrine test is difficult to interpret and sensitivity can be low depending on the underlying genotype and flecainide test results can be inconsistent in a single patient.30
LimitationsThe low number of cases means we cannot extrapolate our conclusions to wider populations. Studies with larger samples and, fundamentally, multicenter studies are needed. Another limitation is the use of flecainide instead of ajmaline to reveal BS; the latter is known to have greater sensitivity. Finally, even when appropriately interpreted, genetic study is not a 100% guarantee of the presence of the disease if a mutation is found that has not previously been described, nor does it rule out disease if genetic variation goes undetected. Hence, given that we excluded patients with probable channelopathy because they had no mutation in the genes sequenced (KCNQ1, KCNH2 and SCN5A principally), we cannot rule out the presence of mutations of other genes such as ryanodine or calsequestrin in patients with negative genotype and, even, in those with a probable pathogenic mutation. Therefore, we consider that the cross sectional design of the study, using the genetic test as the reference for diagnosis, is limited as it excludes patients with a negative genotype in whom the disease cannot be ruled out.
CONCLUSIONSCurrent diagnostic criteria for channelopathies had low sensitivity in our sample of patients and relatives carrying pathogenic mutations. This finding was more noticeable in the group of asymptomatic relatives, who showed very low penetrance, with a high prevalence of normal baseline ECGs. If channelopathy is suspected (IVF, unexplained sudden death or cardiogenic syncope with no apparent heart disease), the combined use of genetic and pharmacological tests in patients and relatives improves the diagnostic protocol's performance in detecting LQTS and BS in patients with a nondiagnostic baseline phenotype.
CONFLICTS OF INTERESTNone declared.