The main cause of morbidity and mortality in Marfan syndrome (MS) and Loeys-Dietz syndrome (LDS) is progressive aortic root dilatation.1,2 Family study is crucial for both early diagnosis and genetic assessment: relatives should be seen by a specialized multidisciplinary team.
We present our experience with the treatment and follow-up of pediatric patients with a diagnosis of MS or LDS. From 2005 to 2016, we followed up 64 pediatric patients in pediatric cardiology: 52 (81%) with classic MS, 2 (3%) with neonatal MS, and 10 (16%) with LDS. Coordinated care involving geneticists, adult cardiologists, ophthalmologists, orthopedic surgeons, and rehabilitation medicine specialists is essential for the comprehensive care of these families.
Fifty-two patients met the criteria for classic MS according to the Ghent criteria.3 Half of these patients (55.8%, 29/52) were investigated due to a known family history, including intrauterine history. The other half (44.2%, 23/52) were diagnosed due to their peculiar phenotype. Of these, 12 (18.8%) were de novo cases with negative genetic studies in both parents; 10 (15.6%) were index cases leading to a diagnosis in another relative. One patient was adopted and therefore the family history was unknown. In summary, 75% had an affected relative, data that coincide with the literature.2
Most of the patients had confirmed FBN1 mutations (80.8%) or a study in-progress (n=2). Seven patients did not undergo genetic testing because they met clinical and family history criteria.
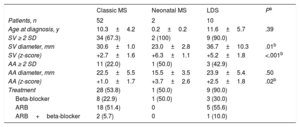
Sixty-seven percent of the patients had dilatation of the sinuses of Valsalva (SV) (Table). This finding agrees with the data in the literature, which describes progressive dilatation in 50% to 83% of pediatric patients.2 The indications for starting treatment were an aortic size adjusted by z-score4 >+2, except for patients younger than 12 years, in whom slight dilatations were tolerated due to the low risk of dissection. Beta-blockers and angiotensin II receptor blockers have been reported as equally effective in adults5 and they were prescribed equally in our patients (Table).
Echocardiographic and Pharmacological Treatment Data
| Classic MS | Neonatal MS | LDS | Pa | |
|---|---|---|---|---|
| Patients, n | 52 | 2 | 10 | |
| Age at diagnosis, y | 10.3±4.2 | 0.2±0.2 | 11.6±5.7 | .39 |
| SV ≥ 2 SD | 34 (67.3) | 2 (100) | 9 (90.0) | |
| SV diameter, mm | 30.6±1.0 | 23.0±2.8 | 36.7±10.3 | .01b |
| SV (z-score) | +2.7±1.6 | +6.3±1.1 | +5.2±1.8 | <.001b |
| AA ≥ 2 SD | 11 (22.0) | 1 (50.0) | 3 (42.9) | |
| AA diameter, mm | 22.5±5.5 | 15.5±3.5 | 23.9±5.4 | .50 |
| AA (z-score) | +1.0±1.7 | +3.7±2.6 | +2.5±1.8 | .02b |
| Treatment | 28 (53.8) | 1 (50.0) | 9 (90.0) | |
| Beta-blocker | 8 (22.9) | 1 (50.0) | 3 (30.0) | |
| ARB | 18 (51.4) | 0 | 5 (55.6) | |
| ARB+beta-blocker | 2 (5.7) | 0 | 1 (10.0) |
AA, ascending aorta; ARB, angiotensin II receptor blocker; LDS, Loeys-Dietz syndrome; MS, Marfan syndrome; SD, standard deviation; SV, sinuses of Valsalva.
Unless otherwise indicated, data are expressed as No. (%) or mean±SD.
No patients died, and 2 required pediatric cardiac surgery (3.8%): 1 adolescent aged 13.3 years required a David procedure for aortic root replacement (SV, 47mm; z-score,+6.3) and 1 child aged 6 years required mitral valve replacement for severe mitral regurgitation.
The 2 patients with neonatal MS were diagnosed at birth and had a rapidly progressive clinical course. Neither had a positive family history. One died at 4.5 months, following mitral valve replacement for severe mitral regurgitation. The other is in the first year of life, with severe aortic root dilatation and moderate-severe mitral and tricuspid regurgitation that is being treated medically. Neonatal MS is uncommon and has a very severe phenotype, with aortic root dilatation in 93% and a mortality reaching 95% in the first year of life.2
The 10 patients with LDS diagnosed in childhood came from 8 families. Two siblings were diagnosed following the sudden death of their mother due to aortic dissection. At 12.5 years, the younger sister died due to aortic dissection (SV, 49mm; z-score,+7.3); she had previously declined a preventative intervention. The older brother underwent a David procedure for aortic root replacement at 14 years of age (SV, 41mm; z-score,+5.4) but died 5 years later due to subarachnoid hemorrhage. In the other 7 families, there were 8 affected individuals: 3 of them were diagnosed because they had affected relatives, 3 were de novo cases, and 2 were index cases leading to diagnosis in their relatives. All the patients had a positive genetic study (4 TGFBR1, 5 TGFBR2) with the exception of the adolescent who died before undergoing genetic study (her bother had a TGFBR1 mutation).
The SV and ascending aorta dilatation was significantly more severe in the patients with LDS than in the patients with MS (Table). Only 1 patient (13 years old) had a normal sized aorta.
All patients with LDS and dilatation (z-score4,6 >+2) received treatment with angiotensin II receptor blockers (Table). Three patients (30%) required cardiac surgery, at 12, 14, and 17 years, in the form of aortic root replacement: 2 underwent a David procedure (SV, 42 and 41mm; z-score,+4.7 and+5.4 respectively) and 1 underwent a Bentall procedure (performed in another hospital). Only 1 patient died before adulthood (the patient who declined intervention).
Loeys-Dietz syndrome confers an increased risk of aortic dissection and cerebral hemorrhage, even in childhood.1 Regular imaging checks of the entire arterial tree are recommended, preferably with magnetic resonance angiography in children.
In conclusion, the follow-up of patients with MS and LDS in specialized hereditary heart disease units is essential for the comprehensive care of these families. In up to 20% of these pediatric patients, follow-up led to the diagnosis of an affected relative, and 55% of the pediatric patients were diagnosed because they had an affected relative. Diagnosis and treatment at an early age can change the natural course of the disease, which can be particularly severe and rapid in neonatal MS and LDS.