Marfan syndrome (MIM 154700) is an autosomal dominant disease affecting the skeleton, eyes, and cardiovascular system. Its estimated prevalence is 2 to 3 per 10 000 individuals. According to the revised Ghent nosology, diagnosis can be established by the presence of a pathogenic mutation in the fibrilin-1 gene (FBN1) in association with aortic root dilatation.1 Of the more than 1800 mutations identified, most are specific to a single family, and 25% are de novo mutations; high intrafamiliar and interfamiliar variation has prevented the establishment of a correlation between genotype and phenotype.1 A possible cause of the varied phenotypic expression is parental mosaicism, which should be borne in mind in genetic counseling after diagnosis of de novo cases. However, there have been few reports of families with Marfan syndrome associated with mosaicism in FBN1.2–5
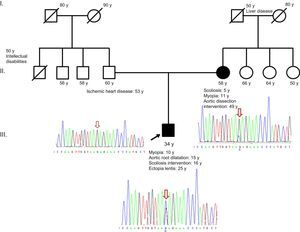
Here, we describe a new mosaic splicing mutation in FBN1. The proband is a 34-year-old man with no siblings. He was diagnosed with Marfan syndrome on the basis of ectopia lentis and aortic root dilatation and has a systemic score of 4 (myopia > 3 diopters, positive thumb sign, chest deformity, and treated scoliosis). Family history includes ischemic heart disease in the father, treated by revascularization, and type B aortic dissection in the mother, treated by percutaneous placement of an endovascular prosthesis (Figure).
. The mutated allele (cytosine, blue) is present in a lower proportion in the mother than in the proband, suggesting mosaicism.")
Following informed consent, the proband underwent a genetic analysis by mass sequencing of 30 genes related to aortic disease. The study identified a previously unknown heterozygous mutation in intron 22 of FBN1 (c.2677+5G>C; NM_000138.4). In silico analysis (with SSF, MaxEnt, NNSplice, and HFF) showed that the mutation disrupts the natural splicing donor site, indicating possible disease association. This variant has not been reported before in the general population (dbSNP, Exome Variant Server); however, a mutation involving a different nucleotide change at the same position has been reported in a Marfan syndrome patient, suggesting that this position is important for correct RNA processing.6 The genetic analysis also identified 2 mutations of unknown pathogenicity, in TGFBR1 (c.409G>A; p.Val137Ile) and in LMNA (c.1158-6C>T, NM_170707.3).
A familial study confirmed that the mother meets the diagnostic criteria for Marfan syndrome (aortic dissection and family history) with a systemic score of 3 (scoliosis, myopia > 3 diopters, and pectus excavatum). She has 3 sisters with no disease phenotype, and both elderly parents died several years ago, apparently from noncardiovascular causes, although no data are available.
The familial cosegregation analysis began with the mother and was directed at FBN1 and at TGFBR1, included because of its association with familial aortic syndromes. The study showed that the mother does not carry the TGFBR1 mutation but is mosaic for the FBN1 mutation (Figure). The presence of this somatic mosaicism indicates that the mutation arose de novo during the mother's embryonic development, and the analysis was therefore not continued in her sisters. Following recommended procedures for confirmation of suspected mosaicism, we extended the genetic analysis to another tissue (bucal mucosa) and confirmed the results with a second independent primer pair. These tests yielded a similar percentage of cells containing the mutant allele, indicating that the mutation event occurred in the early stages of embryogenesis.
The literature on mosaicism in Marfan syndrome reveals that parent carriers have less severe phenotypes than probands or express no manifest phenotype, independently of sex.2–5 However, in the family studied here, the mother has a prominent vascular phenotype, despite carrying the mutation in mosaicism. This discrepancy could in principle reflect differences in the type of genetic alteration; however, this hypothesis is not supported by the published data, which show that a weaker phenotype in the mosaic parent is found with all types of mutation, whether causing amino acid substitution or protein truncation.2–5 We therefore recommend close clinical follow-up of patients with a mosaic mutation, even if it is present in a low percentage of cells.
The intrafamiliar variation encountered here might be explained by a protective effect of the TGFBR1 variant found in the son. Another possible explanation is the age difference, since aortic dilatation in Marfan syndrome is progressive and therefore likely to worsen with age. Moreover, pregnancy is an additional risk factor for aortic dilatation in patients with underlying aortic disease.1
In summary, we present an example of somatic mosaicism in FBN1 that illustrates the importance of considering this possibility in genetic counseling programs. This would permit a more focused strategy than would be possible otherwise. This case also shows that somatic mosaicism is not always associated with a less severe progression of Marfan syndrome.
The authors thank the patient and his family for their cooperation during this study.