Major advances in the field of molecular genetics have expanded our ability to identify genetic substrates underlying the pathogenesis of various disorders that follow Mendelian inheritance patterns. Included among these disorders are the potentially lethal and heritable channelopathies and cardiomyopathies for which the underlying genetic basis has been identified and is now better understood. Clinical and genetic heterogeneity are hallmark features of these disorders, with thousands of gene mutations being implicated within these divergent cardiovascular diseases. Genetic testing for several of these heritable channelopathies and cardiomyopathies has matured from discovery to research-based genetic testing to clinically/commercially available diagnostic tests. The purpose of this review is to provide the reader with a basic understanding of human medical genetics and genetic testing in the context of cardiovascular diseases of the heart. We review the state of clinical genetic testing for the more common channelopathies and cardiomyopathies, discuss some of the pertinent issues that arise from genetic testing, and discuss the future of personalized medicine in cardiovascular disease.
Keywords
.
INTRODUCTIONConsiderable advances in the field of molecular genetics have provided important tools to elucidate genetic substrates for many genetic disorders that follow Mendelian inheritance patterns. The genetic underpinnings of heritable and potentially lethal cardiomyopathies and cardiac channelopathies including hypertrophic cardiomyopathy (HCM), dilated cardiomyopathy (DCM), arrhythmogenic right ventricular cardiomyopathy, long QT syndrome (LQTS), catecholaminergic polymorphic ventricular tachycardia (CPVT), and Brugada syndrome (BS) have been identified and are now better understood.
Marked clinical and genetic heterogeneity are cardinal characteristics of these disorders, with multiple genes underlying their pathogenic mechanisms. Genetic testing for several of these heritable cardiomyopathies and channelopathies has made the transition from research-based discovery to clinical utility and are now commercially available as clinical genetic tests.
In this review, we will assess the state of clinical genetic testing for the most common channelopathies/cardiomyopathies: LQTS, CPVT, BS, and HCM. Next, we will discuss the need for careful interpretation of genetic test results, the importance of the genetic counselor, and the future of personalized medicine in cardiovascular disease.
GENETIC TESTING FOR INHERITED CARDIAC CHANNELOPATHIES AND CARDIOMYOPATHIESAlthough genetic testing for DCM and arrhythmogenic right ventricular cardiomyopathy is evolving rapidly and is commercially available, interpreting the test results is extremely challenging due to unclear clinical impact apart from first degree relative confirmatory mutation testing, lower yields, and comparatively high “background noise rates”1,2. This review will focus on LQTS, CPVT, BS, and HCM, where the clinical utility of genetic testing is arguably the greatest at present. In each of the following disease sections, we will provide a brief clinical description, the genetic basis for the disorder, and the current Heart Rhythm Society (HRS)/European Heart Rhythm Association (EHRA) expert consensus recommendations for genetic testing.3
Long QT SyndromeClinical DescriptionLQTS is characterized by delayed repolarization of the myocardium, QT prolongation, and T wave abnormalities on a resting 12-lead surface electrocardiogram (ECG) that can manifest as syncope, seizures, and sudden cardiac death in patients with a structurally normal heart.4 Although the prevalence of LQTS is estimated to be as high as 1:2500 individuals, due to the genetic heterogeneity of this disease individuals with LQTS may be missed and may not present with QT prolongation on a resting 12-lead ECG.5 The typical manifestation of syncope, seizures, and sudden cardiac death usually occurs following adrenergic stimulation (auditory stimuli, exercise, emotion, etc.) or during the post partum period.6 The hallmark arrhythmia of torsade de pointes often spontaneously returns to normal sinus rhythm, resulting in only an episode of syncope; however, approximately 5% of untreated and unsuspecting LQTS individuals succumb to a fatal arrhythmia as their first event. Many of these individuals may have had a previous unrecognized cardiac event consistent with the LQTS phenotype. In addition, approximately 20% of autopsy-negative, sudden, unexplained deaths in the young and 10% of sudden infant death syndrome may be LQTS-triggered deaths.7,8
Genetic BasisLQTS, previously described as Romano-Ward syndrome, is a genetically heterogeneous disorder most often inherited in an autosomal dominant manner. The recessive form of LQTS, previously described as Jervell and Lange-Nielson syndrome, is characterized by a severe cardiac phenotype and sensorineural hearing loss. Hundreds of mutations have now been identified in 10 LQTS-susceptibility genes (Table 1) responsible for a nonsyndromic “classical” LQTS phenotype. In addition, three rare, multisystem disorders—ankyrin-B syndrome, Anderson-Tawil syndrome, and Timothy syndrome—have been annotated previously as LQT4, LQT7, and LQT8, respectively.9–11 Rare de novo, rather than familial, germline mutations have also been described that account for approximately 5% to 10% of LQTS.
Approximately 75% of LQTS is due to mutations in three genes—KCNQ1 (LQT1), KCNH2 (LQT2), and SCN5A (LQT3)—that encode for either potassium (Kv7.1 and Kv11.1) or sodium (Nav1.5) ion channel pore-forming α-subunits that critically orchestrate the cardiac action potential of the ventricular myocytes.12,13 The remainder of genotype-positive LQTS is attributed to mutations in genes that encode other cardiac ion channels or ion channel interacting proteins that regulate ion channel function. The 7 minor LQTS-susceptibility genes contribute <5% of LQTS.
To date, the majority of mutations described in LQTS are single nucleotide substitutions and small insertion/deletions within the coding regions of the LQTS-susceptibility genes. However, a few examples of large gene rearrangements resulting in large gene deletions/duplications have also been described in LQTS.14 While much progress has been made in understanding the underlying etiology of LQTS with 10 genes already described, it is important to understand that approximately 20% to 25% of LQTS remain genetically elusive. With the recent advances in molecular approaches, continued investigation of phenotype-strong, genotype-negative patients offers the greatest probability of identifying the genetic underpinnings of the remaining 20% to 25% of LQTS.
A few genotype-phenotype correlations have been reported in patients with LQT1, LQT2, and LQT3 that may guide genetic testing for LQTS. In LQT1, cardiac events are often seen most frequently during exercise and swimming, whereas in LQT2 and LQT3 cardiac events occur most frequently during periods of rest/sleep. Events due to auditory stimulus and susceptibility of women in their post partum period are characteristics of LQT2.6,15 However, when exercise-induced syncope occurs in a patient with a normal QTc (<440ms in men and <460ms in women) this phenotype may be indicative of CPVT.16
Gene-suggestive ECG patterns have also been described6,15 that may be helpful in guiding the genetic test. For example, broad-based T waves are associated with LQT1, low amplitude notched or biphasic T waves with LQT2, and a long isoelectric segment followed by a narrow-based T wave is associated with LQT3. However, these gene-suggestive T-wave patterns are only suggestive of the LQTS subtype and should not be used to make a pre-genetic test prediction. This cautious approach is of utmost importance as the underlying genetic basis not only has diagnostic implications but can heavily influence the therapeutic recommendations in LQTS. An example is seen in the response of LQTS patients to beta blockers, the standard LQTS pharmacotherapy. We have observed that beta blockers are most protective in LQT1 patients, moderately protective in LQT2 patients, and possibly not sufficiently protective in LQT3 patients.17,18 However, targeting the late sodium current characteristic of LQT3 mutations with agents such as mexiletine, flecainide, ranolazine, or propranolol may be a more gene-specific therapy for LQT3 patients.6,15,19,20
Intra-genotype risk stratification has also been conducted for LQT1 and LQT2 based upon mutation type, mutation location, and cellular function.21,22 Patients with LQT1 missense mutations localizing to the transmembrane region of the Kv7.1 potassium channel have twice the risk of a LQT1-triggered cardiac event than LQT1 patients with mutations localizing to the C-terminus. In addition, cellular in vitro studies of LQT1 patient mutations that resulted in a greater loss-of-function phenotype (ie, dominant negative) show twice the clinical risk compared to mutations that have a less severe impact on Kv7.1 channel biology (ie, haploinsufficiency). Like the molecular risk stratification seen in patients with LQT1, patients with LQT2 also have independent risk factors that are linked to mutation location and cellular function. LQT2 patients with Kv11.1 pore region mutations tend to have a longer QTc, experience a greater number of arrhythmia-related cardiac events at a younger age, and have a more severe clinical manifestation of the disorder compared to LQT2 patients with mutations that do not localize to the pore region of the Kv11.1 channel. Furthermore, recent studies have shown that LQT2 Kv11.1 mutations involving the transmembrane pore region had the greatest risk for cardiac events, those with frame-shift/nonsense mutations in any region had an intermediate risk, and those with missense mutations in the carboxy-terminal region had the lowest risk for cardiac events.22 In 2012, LQTS genotyping fully satisfies the triad in terms of diagnostic (move towards/move away from diagnosis in index case, gold standard rule in/rule out in family members), prognostic (both between and within genotypes), and therapeutic impact (LQT1 patients approached with a fundamentally distinct treatment plan compared to LQT3 patients, for example).
Genetic Testing Recommendations for Long QT SyndromeAny patient with a suspected clinical LQTS diagnosis should be offered clinical genetic testing. As described earlier, genotyping provides additional risk stratification that can guide therapeutic options for the clinician. Genetic testing of the index case currently has a 75% likelihood of identifying the possible/probable LQTS-associated mutation. If the index case has a positive genetic test result, genotyping immediate family members will further refine the genetic diagnosis regardless of symptoms or corrected QT interval. Additionally, patients with unexplained, exertional syncope or drug-induced QT prolongation/torsade de pointes who do not meet full LQTS diagnostic criteria and receive a diagnosis of “possible” LQTS may also benefit from LQTS genetic testing. For these cases, a positive genetic test may be valuable in upgrading the diagnosis from clinically “possible” to genetically probable. In contrast, a negative genetic test would successfully discount 75% of congenital LQTS and provide an independent measure to help the clinician move away from this diagnosis, especially if the clinical phenotype is weak.
Accordingly, the HRS/EHRA expert consensus recommendations3 for LQTS genetic testing are as follows: a) comprehensive or LQT1-3 (KCNQ1, KCNH2, and SCN5A) targeted LQTS genetic testing is recommended for any patient in whom a cardiologist has established a strong clinical index of suspicion for LQTS based on examination of the patient's clinical history, family history, and expressed electrocardiographic (resting 12-lead ECGs and/or provocative stress testing with exercise or catecholamine infusion) phenotype; b) comprehensive or LQT1-3 (KCNQ1, KCNH2, and SCN5A) targeted LQTS genetic testing is recommended for any asymptomatic patient with QT prolongation in the absence of other clinical conditions which might prolong the QT interval (such as electrolyte abnormalities, hypertrophy, bundle branch block, etc.; ie, otherwise idiopathic) on serial 12-lead ECGs defined as QTc>480ms (prepuberty) or >500ms (adults); c) comprehensive or LQT1-3 (KCNQ1, KCNH2, and SCN5A) targeted LQTS genetic testing may be considered for any asymptomatic patient with otherwise idiopathic QTc values>460ms (prepuberty) or >480ms (adults) on serial 12-lead ECGs, and d) mutation-specific genetic testing is recommended for family members and other appropriate relatives subsequently following the identification of the LQTS-causative mutation in an index case.
Catecholaminergic Polymorphic Ventricular TachycardiaClinical DescriptionCPVT is a potentially lethal heritable arrhythmia syndrome, mainly expressed in the young, that usually presents with syncope, seizures, or sudden death associated with exercise. Once thought to manifest only during childhood, recent studies have suggested that this syndrome may first present at any age, from infancy to 40 years. Like LQT1, swimming is a typical trigger for an arrhythmogenic event in CPVT. In fact, both CPVT and LQT1 are responsible for many near-drowning or unexplained drowning events in otherwise healthy swimmers.23 However, contrary to LQTS, CPVT is associated with a completely normal resting 12-lead ECG (although sometimes with bradycardia and U waves), but may present with significant ventricular ectopy, including bidirectional VT after either exercise or catecholamine stress testing.24 Mortality rates of 30% to 50% by age 35 years and the presence of a positive family history of young (<40 years) sudden cardiac death for more than a third of CPVT patients and as many as 60% of families harboring RyR2 mutations illustrate the lethality of CPVT.25 In addition, CPVT is the underlying pathogenic basis for approximately 15% of autopsy-negative sudden unexplained death in the young.8
Genetic BasisCPVT is usually inherited as an autosomal dominant disorder. Alterations in intracellular calcium release from the sarcoplasmic reticulum underlie the pathogenesis of CPVT. RYR2-encoded cardiac ryanodine receptor gain-of-function mutations account for the majority (∼60%) of CPVT. These RyR2 mutations result in excessive diastolic release of calcium from the sarcoplasmic reticulum that can lead to delayed after-depolarizations and ventricular arrhythmias.25 The vast majority (90%) of RYR2 mutations are missense mutations; however, as much as 5% of unrelated CPVT patients host large RYR2 gene arrangements that result in large exon deletions. Two recessive forms of CPVT have been identified involving mutations in CASQ2-encoded calsequestrin-2 protein and TRDN-encoded triadin, two RYR2 interacting proteins.26,27 Most recently, mutations in calmodulin-1 (CALM1) have been identified as a cause of autosomal dominant CPVT, with the elucidation of a single missense mutation that segregated with a CPVT phenotype in a large Swedish pedigree (Table 1).28
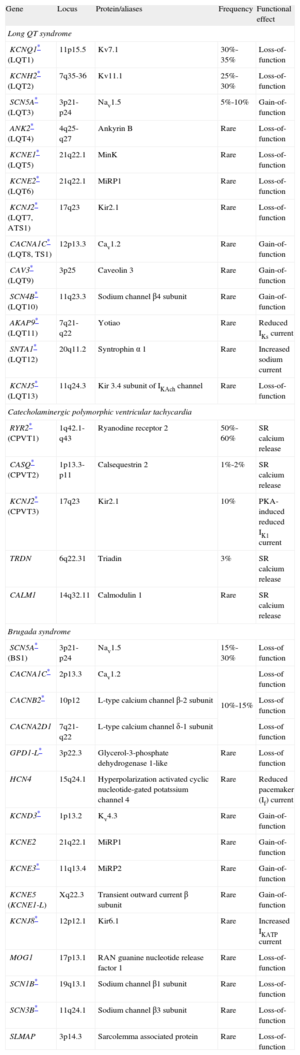
Summary of Genes Associated With Inherited Cardiac Channelopathies.
| Gene | Locus | Protein/aliases | Frequency | Functional effect |
| Long QT syndrome | ||||
| KCNQ1* (LQT1) | 11p15.5 | Kv7.1 | 30%-35% | Loss-of-function |
| KCNH2* (LQT2) | 7q35-36 | Kv11.1 | 25%-30% | Loss-of-function |
| SCN5A* (LQT3) | 3p21-p24 | Nav1.5 | 5%-10% | Gain-of-function |
| ANK2* (LQT4) | 4q25-q27 | Ankyrin B | Rare | Loss-of-function |
| KCNE1* (LQT5) | 21q22.1 | MinK | Rare | Loss-of-function |
| KCNE2* (LQT6) | 21q22.1 | MiRP1 | Rare | Loss-of-function |
| KCNJ2* (LQT7, ATS1) | 17q23 | Kir2.1 | Rare | Loss-of-function |
| CACNA1C* (LQT8, TS1) | 12p13.3 | Cav1.2 | Rare | Gain-of-function |
| CAV3* (LQT9) | 3p25 | Caveolin 3 | Rare | Gain-of-function |
| SCN4B* (LQT10) | 11q23.3 | Sodium channel β4 subunit | Rare | Gain-of-function |
| AKAP9* (LQT11) | 7q21-q22 | Yotiao | Rare | Reduced IKs current |
| SNTA1* (LQT12) | 20q11.2 | Syntrophin α 1 | Rare | Increased sodium current |
| KCNJ5* (LQT13) | 11q24.3 | Kir 3.4 subunit of IKAch channel | Rare | Loss-of-function |
| Catecholaminergic polymorphic ventricular tachycardia | ||||
| RYR2* (CPVT1) | 1q42.1-q43 | Ryanodine receptor 2 | 50%-60% | SR calcium release |
| CASQ* (CPVT2) | 1p13.3-p11 | Calsequestrin 2 | 1%-2% | SR calcium release |
| KCNJ2* (CPVT3) | 17q23 | Kir2.1 | 10% | PKA-induced reduced IK1 current |
| TRDN | 6q22.31 | Triadin | 3% | SR calcium release |
| CALM1 | 14q32.11 | Calmodulin 1 | Rare | SR calcium release |
| Brugada syndrome | ||||
| SCN5A* (BS1) | 3p21-p24 | Nav1.5 | 15%-30% | Loss-of function |
| CACNA1C* | 2p13.3 | Cav1.2 | 10%-15% | Loss-of function |
| CACNB2* | 10p12 | L-type calcium channel β-2 subunit | Loss-of function | |
| CACNA2D1 | 7q21-q22 | L-type calcium channel δ-1 subunit | Loss-of function | |
| GPD1-L* | 3p22.3 | Glycerol-3-phosphate dehydrogenase 1-like | Rare | Loss-of function |
| HCN4 | 15q24.1 | Hyperpolarization activated cyclic nucleotide-gated potatssium channel 4 | Rare | Reduced pacemaker (If) current |
| KCND3* | 1p13.2 | Kv4.3 | Rare | Gain-of-function |
| KCNE2 | 21q22.1 | MiRP1 | Rare | Gain-of-function |
| KCNE3* | 11q13.4 | MiRP2 | Rare | Gain-of-function |
| KCNE5 (KCNE1-L) | Xq22.3 | Transient outward current β subunit | Rare | Gain-of-function |
| KCNJ8* | 12p12.1 | Kir6.1 | Rare | Increased IKATP current |
| MOG1 | 17p13.1 | RAN guanine nucleotide release factor 1 | Rare | Loss-of-function |
| SCN1B* | 19q13.1 | Sodium channel β1 subunit | Rare | Loss-of-function |
| SCN3B* | 11q24.1 | Sodium channel β3 subunit | Rare | Loss-of-function |
| SLMAP | 3p14.3 | Sarcolemma associated protein | Rare | Loss-of-function |
ATS, Anderson-Tawil syndrome; BS, Brugada syndrome; CPVT, catecholaminergic polymorphic ventricular tachycardia; LQT: long QT; SR, sarcoplasmic reticulum; TS, Timothy syndrome.
Importantly, nearly a third of “possible/atypical” LQTS (QTc<480 ms) cases with exercise-related syncope or cardiac arrest have been identified as RYR2 mutation positive. Thus, a clinical presentation of exertion-induced cardiac events (ie, syncope, seizures, and/or cardiac arrest) and a QTc<460ms should always encourage CPVT as a first clinical consideration rather than “concealed LQT1”. In fact, as much as 30% of CPVT patients may have been misdiagnosed as “LQTS with normal QT intervals” or “concealed LQTS”, illustrating the critical clinical significance of properly differentiating between CPVT and LQTS, as risk assessments and treatment strategies of these distinct disorders may vary. Similarly, some CPVT-diagnosed patients with the presence of bi-directional VT on exercise have been shown to harbor KCNJ2 mutations linked to the rarely lethal Andersen-Tawil syndrome type 1.29 The misdiagnosis of Anderson-Tawil syndrome as the highly lethal disorder CPVT could result in a more aggressive prophylactic therapy (ie, implantable cardioverter defibrillator implantation) than necessary. Genetic testing may provide a clear diagnostic differentiation between CPVT and concealed LQT1 and between CPVT and Anderson-Tawil syndrome type 1.
Genetic Testing Recommendations for Catecholaminergic Polymorphic Ventricular TachycardiaThe HRS/EHRA expert consensus recommendations3 for CPVT genetic testing state the following: a) comprehensive or CPVT1 and CVPT2 (RYR2 and CASQ2) targeted CPVT genetic testing is recommended for any patient in whom a cardiologist has established a clinical index of suspicion for CPVT based on examination of the patient's clinical history, family history, and expressed electrocardiographic phenotype during provocative stress testing with cycle, treadmill, or catecholamine infusion, and b) mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the CPVT-causative mutation in an index case. In addition, those patients with exercise-induced syncope/cardiac arrest or near-drowning in the setting of a QTc<460ms should be considered for CPVT genetic testing.
Brugada SyndromeClinical DescriptionBS is a potentially lethal heritable syndrome characterized by a coved-type ST-segment elevation (≥2mm) followed by a negative T-wave recorded in the right precordial leads V1 through V3 (“type 1” BS ECG pattern), and an increased predisposition for sudden death resulting from episodes of polymorphic ventricular tachyarrhythmias. A “type 2” ECG pattern consisting of a saddle-back ST-segment morphology ≥2mm followed by a positive T-wave, and a “type 3” ECG pattern consisting of either a coved or saddle-back type ST-segment elevation of ≤1mm have also been observed but are not considered diagnostic for BS. Although BS can manifest at any age, it predominantly affects young male adults (30-40 years of age). Symptoms including sudden cardiac death usually occur during sleep.30
Genetic BasisBS is typically inherited in an autosomal dominant fashion although up to half of all cases may be sporadic in nature. To date 15 BS-susceptibility genes (Table 1) have been identified. Loss-of-function mutations in the SCN5A-encoded α-subunit of the cardiac sodium channel represent the most common genetic substrate for BS, accounting for 15% to 30% of the disorder.31 Besides sodium channel dysfunction, mutations involving the L-type calcium channel α1 (CACNA1C32), β2 (CACNB2B32), and α2δ (CACNA2D133) subunits may cause 10% to 15% of BS, especially when accompanied by short QT intervals.33 However, in 2012, Crotti et al. performed the first comprehensive mutational analysis in a large cohort of unrelated BS patients. Although they identified SCN5A mutations in 16% of their cohort, only 1.5% of their BS cases had a mutation in one of the three L-type calcium channel subunit genes, in the absence of a short QT interval. In fact, all of the minor BS genes account for less than 5% of the disorder.31 Thus it is important to keep in mind that as of 2012, approximately 70% to 80% of BS remains genetically elusive.
While somewhat limited, some key genotype/phenotype correlations derived from research based genetic testing of BS patients may be helpful in directing future genetic testing efforts and provide some prognostic utility of BS genetic testing. For example, the presence of a long PQ interval (≥200ms) on ECG is more suggestive of SCN5A-mediated BS.34 In fact, Crotti et al. reported that compared to a <10% yield for a positive SCN5A genetic test for those patients with a PQ<200ms, the yield was almost 40% among patients with a PQ interval≥200ms. Furthermore, the presence of a short QT interval (QTc <350ms) points toward an L-type calcium channel-mediated BS substrate.31
In 2009, Meregalli et al. investigated whether the type of SCN5A mutation correlated with the severity of conduction slowing present in overlapping aberrant rhythm phenotypes of progressive cardiac conduction disease and BS.35 Their results revealed that patients with mutations that resulted in a more severe reduction in peak sodium current had a more severe phenotype of syncope and conduction defect, providing the first report of intra-genotype risk stratification associated with loss-of-function sodium channelopathies.
Genetic Testing Recommendations for Brugada SyndromePatients with suspected BS could undergo clinical genetic testing as long as it is recognized that the yield from currently available BS genetic tests is approximately 20% to 30%. Further, except for the recent association between SCN5A mutation type and clinical severity, the primary role of BS genetic testing is diagnostic confirmation of root cause for an index case. This is followed by confirmatory genetic testing of immediate family members to distinguish those needing ongoing clinical surveillance and preventive measures, such as avoiding certain drugs (www.brugadadrugs.org)36, from those who can be dismissed as clinically, electrocardiographically, and genetically unaffected.
The HRS/EHRA expert consensus recommendations3 for BS genetic testing are the following: a) comprehensive or BS1 (SCN5A) targeted BS genetic testing can be useful for any patient in whom a cardiologist has established a clinical index of suspicion for BS based on examination of the patient's clinical history, family history, and expressed electrocardiographic (resting 12-lead ECGs and/or provocative drug challenge testing) phenotype; b) genetic testing is not indicated in the setting of an isolated type 2 or type 3 Brugada ECG pattern, and c) Mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the BS-causative mutation in an index case.
Hypertrophic CardiomyopathyClinical DescriptionHCM is regarded as asymmetrical left ventricular hypertrophy without a clinically identifiable cause, has an estimated prevalence of 1:500 people, and is a common cause of sudden cardiac death in the young, particularly in the young athlete. The expressivity of HCM varies greatly, ranging from an asymptomatic lifelong clinical course to chest pain, exertion-related dyspnea, syncope, progressive exercise intolerance, or sudden death as the first event, which may occur at any age.37,38 The morphological phenotype in HCM spans from minuscule to extensive hypertrophy, very little to extreme fibrosis and myocyte disarray, absent to severe left ventricular outflow tract obstruction and unique anatomical subtypes that include reverse curve, sigmoidal, and apical variant HCM.39
Genetic BasisHCM is typically inherited in an autosomal dominant manner. At least 27 putative HCM-susceptibility genes have been implicated to date, with hundreds of mutations identified (Table 2). Sarcomeric or myofilament HCM is the most common HCM genetic subtype, due to mutations in genes encoding for proteins of the thick and thin myofilaments of the cardiac sarcomere (Table 2). The two most common HCM-associated genes, MYBPC3 and MYH7, have an estimated prevalence of 25% to 35% for each gene and account for the majority of research-based positive genetic tests. In addition, cardiac Z-disc, calcium (Ca2+)—handling, and regulatory protein-encoding genes have been linked to HCM pathogenesis. Mutations in several cellular metabolic-related protein-encoding genes result in unique disorders associated with left ventricular hypertrophy that can simulate the HCM phenotype.
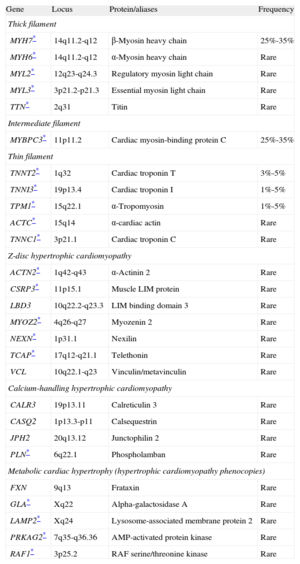
Summary of Genes Associated With Hypertrophic Cardiomyopathy.
| Gene | Locus | Protein/aliases | Frequency |
| Thick filament | |||
| MYH7* | 14q11.2-q12 | β-Myosin heavy chain | 25%-35% |
| MYH6* | 14q11.2-q12 | α-Myosin heavy chain | Rare |
| MYL2* | 12q23-q24.3 | Regulatory myosin light chain | Rare |
| MYL3* | 3p21.2-p21.3 | Essential myosin light chain | Rare |
| TTN* | 2q31 | Titin | Rare |
| Intermediate filament | |||
| MYBPC3* | 11p11.2 | Cardiac myosin-binding protein C | 25%-35% |
| Thin filament | |||
| TNNT2* | 1q32 | Cardiac troponin T | 3%-5% |
| TNNI3* | 19p13.4 | Cardiac troponin I | 1%-5% |
| TPM1* | 15q22.1 | α-Tropomyosin | 1%-5% |
| ACTC* | 15q14 | α-cardiac actin | Rare |
| TNNC1* | 3p21.1 | Cardiac troponin C | Rare |
| Z-disc hypertrophic cardiomyopathy | |||
| ACTN2* | 1q42-q43 | α-Actinin 2 | Rare |
| CSRP3* | 11p15.1 | Muscle LIM protein | Rare |
| LBD3 | 10q22.2-q23.3 | LIM binding domain 3 | Rare |
| MYOZ2* | 4q26-q27 | Myozenin 2 | Rare |
| NEXN* | 1p31.1 | Nexilin | Rare |
| TCAP* | 17q12-q21.1 | Telethonin | Rare |
| VCL | 10q22.1-q23 | Vinculin/metavinculin | Rare |
| Calcium-handling hypertrophic cardiomyopathy | |||
| CALR3 | 19p13.11 | Calreticulin 3 | Rare |
| CASQ2 | 1p13.3-p11 | Calsequestrin | Rare |
| JPH2 | 20q13.12 | Junctophilin 2 | Rare |
| PLN* | 6q22.1 | Phospholamban | Rare |
| Metabolic cardiac hypertrophy (hypertrophic cardiomyopathy phenocopies) | |||
| FXN | 9q13 | Frataxin | Rare |
| GLA* | Xq22 | Alpha-galactosidase A | Rare |
| LAMP2* | Xq24 | Lysosome-associated membrane protein 2 | Rare |
| PRKAG2* | 7q35-q36.36 | AMP-activated protein kinase | Rare |
| RAF1* | 3p25.2 | RAF serine/threonine kinase | Rare |
Genetic testing of the 8 or 9 established HCM-susceptibility genes for sarcomeric HCM provides a mutation detection yield ranging from 35% to 65% among several different international research cohorts of unrelated patients who met the clinical criterion of HCM.37,40 Additionally, echo-guided genetic testing may assist the clinician by providing an estimate of the a priori probability of a positive genetic test based upon morphologic subtyping following echocardiography.41
HCM genetic testing contributes primarily to the diagnostic confirmation, with only a minor role in terms of prognosis and therapeutic impact. Genetic testing for HCM may play a crucial role in differentiating true HCM from exercise-adaptive hypertrophy (“athlete's heart”) or from HCM phenotypic mimickers such as Anderson-Fabry, glycogen (PRKAG2) and lysosomal (LAMP2) storage, mitochondrial, Noonan and LEOPARD syndromes, for which there are definitive and alternative therapies distinct from HCM treatments. However, a negative genetic test cannot “rule out” HCM. Further, prophylactic therapies may potentially prevent or delay the onset of hypertrophy among those patients who are genotype positive but currently hypertrophy negative.
One must exercise great restraint when considering a prophylactic implantable cardioverter defibrillator decision based solely upon the genetic test result. Currently, clinical data supports a “mutation class effect” prognostic indicator whereby, compared to clinically diagnosed HCM patients with a negative genetic test, unrelated patients with a positive sarcomeric HCM genetic test express more severe hypertrophy, a younger age at diagnosis, and a higher probability of proceeding towards end-stage disease.42 However, it is unclear how this observation may translate to a therapeutic approach for the patient with sarcomeric/myofilament HCM (ie, a positive genetic test) compared to the patient with negative genetic test HCM.
Genetic Testing Recommendations for Hypertrophic CardiomyopathyGenetic testing for the HCM patient may provide a diagnostic standard for their relatives. A positive genetic test would allow for the systematic analysis of an HCM-affected proband's relatives to identify those family members who are mutation positive regardless of their current clinical phenotypic manifestation, allowing for early detection and appropriate selection of relatives for continued clinical surveillance while dismissing those family members who are mutation negative/phenotype negative from regular cardiac evaluations and echocardiograms.
Accordingly, the HRS/EHRA expert consensus recommendations3 for HCM genetic testing state: a) comprehensive or targeted (MYBPC3, MYH7, TNNI3, TNNT2, TPM1) HCM genetic testing is recommended for any patient in whom a cardiologist has established a clinical diagnosis of HCM based on examination of the patient's clinical history, family history, and electrocardiographic/echocardiographic phenotype, and b) mutation-specific genetic testing is recommended for family members and appropriate relatives following the identification of the HCM-causative mutation in an index case.
INTERPRETATION OF GENETIC TEST RESULTSThe clinical management and evaluation of patients and families suspected of having a potentially lethal cardiac disorder should be carried out under the supervision of a cardiologist with specific expertise in heritable channelopathies and cardiomyopathies.43 Due to the incomplete penetrance and variable expressivity associated with many of these disorders, interpretation of the genetic test result must be done cautiously, with careful consideration given to the assignment of pathogenicity to the identified genetic variant. To illustrate, in contrast to rare pathogenic LQTS-associated mutations present in an estimated 1:2500 persons (0.04%) and 75% of LQTS patients with a robust clinical phenotype, comprehensive mutational analysis of the three major LQTS genes KCNQ1 (LQT1), KCNH2 (LQT2) and SCN5A (LQT3) for over 1300 otherwise healthy volunteers revealed that approximately 4% of Caucasians and as many as 8% of non-Caucasians host rare (<0.5% allelic frequency) amino acid-altering (nonsynonymous) genetic variants in these cardiac channel genes.44 While some of the variants identified in this ostensibly healthy population may represent subclinical disease modifiers, the vast majority must represent benign background “genetic noise”. Thus, when performed in Caucasian cases with a robust phenotype compared to controls, LQTS genetic testing has a “signal-to-noise” ratio of approximately 19:1. This observation of background nonpathogenic missense variants is not confined to LQTS, but likely extends to all of the heritable cardiomyopathies and cardiac channelopathies detailed in this review in varying degrees. Therefore, it is extremely important to recognize that cardiomyopathy/channelopathy genetic tests are probabilistic tests rather than binary (yes or no) tests. In order to differentiate pathogenic disease-causing mutations from otherwise rare variants of uncertain significance, variant interpretation algorithms are beginning to emerge for each of these disorders.2,44.
GENETIC COUNSELINGGenetic counseling is an essential component of the genetic testing process. Given the complexity of medical genetic information, it is advantageous for the ordering physician to work closely with a masters-trained, board-certified genetic counselor, preferably one with specialized training in cardiovascular genetics, as a team that will communicate with the patient regarding the implications of genetic testing and the benefits and consequences of testing. Counseling should continue throughout all phases of testing in order to provide the patient with the necessary information and resources to understand, cope with, and make decisions about genetic disease.45 A genetic counselor may be useful in many aspects and can assist in obtaining a complete multigenerational family history and providing information on the disorder, its mode of inheritance, and its implications for future family planning. In addition, it is important for the genetic counselor to explain the genetic test itself, the possible limitations, costs, and potential for unforeseen results, as well as the potential impact of the genetic test result on the patient and family members.37 The National Society of Genetic Counselors (available at: www.nsgc.org)46 is a useful resource for clinicians seeking a genetic counselor with experience in a specific disease.
FUTURE DIRECTIONS IN CARDIOVASCULAR GENETIC DIAGNOSISNext-Generation Whole Exome/Genome Sequencing for Routine Genetic Diagnosis and New Disease Gene DiscoveryThe emergence of next-generation sequencing platforms that have substantially decreased the cost of genotyping has now provided new tools to efficiently interrogate an individual's entire genome or exome (entire amino acid-encoding region of the genome) in a single reaction. Rather than a “one gene at a time” approach to genetic testing, specialized genomics laboratories around the world are now beginning to offer whole genome or whole exome sequencing of a patient's DNA sample. This technology effectively provides a list of every single nucleotide substitution and small insertion/deletion (common or rare, benign or pathogenic) for every gene in a patient's genome.
Whole genome and whole exome sequencing using next-generation sequencing have identified novel genes for several Mendelian disorders.47 Proof of this concept was first illustrated by Ng et al. in 2009, who used whole exome sequencing of 4 unrelated individuals affected with the rare disorder Freeman-Sheldon syndrome and 8 controls to identify the single candidate gene MYH3 for this Mendelian disorder.48 Further studies using both whole exome and whole genome have also identified novel candidate genes in several other disorders such as Kabuki syndrome,49 autosomal recessive dilated cardiomyopathy,50 and Charcot-Marie-Tooth disease51 proving the usefulness of these next-generation sequencing strategies.
Genetic Modifiers of Cardiovascular DiseasesThe quest to identify genetic modifiers that explain the reduced penetrance and variable expressivity often observed among unrelated individuals and even within pedigrees for these potentially lethal cardiac channelopathies and cardiomyopathies will be at the forefront of cardiovascular genetics research for the foreseeable future.
Genome Wide Association StudiesThe completion of the final draft of the Human Genome Project in 2003 and the emergence of high-throughput genotyping methods has provided the tools to perform large population-based genome-wide association studies (GWAS). These studies involve sequencing of thousands of individuals to access very common single nucleotide polymorphisms (SNPs) that are associated with complex (non-Mendelian or polygenic) traits. Several GWAS have now been performed, identifying thousands of SNPs reportedly associated with various complex diseases or traits listed in the Catalog of Published GWAS (www.genome.gov/26525384).52 Recently, GWAS have been performed to identify SNPs associated with cardiovascular risk factors predisposing to coronary artery disease,53 as well as electrocardiographic traits consistent with increased risk of ventricular arrhythmias and sudden cardiac death, including the PR interval, QRS duration, and QT interval.54–56 A meta-analysis of these studies revealed the strongest association between variants in the NOS1AP (capon) gene with QT interval duration, and follow-up studies have highlighted the importance of the nitric oxide synthase pathway in myocardial function and action potential repolarization.57,58 Continued refinement of GWAS may hold great promise in identifying key genetic contributors that may explain in some part the reduced penetrance and variable expressivity often observed in the cardiac channelopathies and cardiomyopathies discussed here.
MicroRNAs as Potential Contributors and Modifiers of Cardiovascular DiseaseMicroRNAs may also underlie cardiac disease susceptibility and phenotypic variation. MicroRNAs are post-transcriptional repressors of gene expression that bind to the 3′ untranslated region of target gene transcripts to eliminate microRNAs that should not be expressed at a particular time or in a particular cell type. This mechanism is hypothesized to contribute to the individual disease heterogeneity observed. To date, several studies have revealed that microRNAs binding site polymorphisms can either cause or modulate many disease phenotypes, such as asthma59 and Friedrich ataxia,60 and more recently the dysregulation of specific microRNAs has been linked to the development and modulation of diseases of the heart.61,62 For example, study results using microRNA expression arrays show differential regulation of microRNAs in various diseases—including HCM, dilated cardiomyopathy, and ischemic cardiomyopathy—compared to normal hearts.63 Most recently, Amin et al. elegantly demonstrated how microRNA binding site polymorphisms in the 3′ untranslated region of KCNQ1-encoded Kv7.1 potassium channels serve as a potential allele-specific modifier of QT interval and symptoms in patients with LQT1, exceeding any other genetic modifier effect.64 Their study identified 3 naturally occurring single nucleotide polymorphisms within the 3′ untranslated region of KCNQ1 whereby the presence of the minor allele haplotype on the normal allele accentuated the LQT1 phenotype by suppressing healthy KCNQ1 transcripts, while the presence of the minor allele haplotype on the mutant allele attenuated the LQT1 phenotype by suppressing mutant KCNQ1 transcripts. These findings not only illustrate a new concept in our understanding of disease-modifying genetic determinants of Mendelian diseases, but also may explain a significant portion of the variable expressivity and reduced penetrance observed in the cardiac channelopathies.65
Induced Pluripotent Stem Cell-Derived CardiomyocytesRecent advances in cellular programming and reprogramming have provided new avenues for understanding the etiology of complex diseases. The ground-breaking and Nobel prize-winning discovery by Drs. Takahashi and Yamanaka that mouse embryonic and adult fibroblasts could acquire embryonic stem cell-like characteristics following retroviral transduction with 4 transcription factors (Oct3/4, Sox2, Klf4, and c-Myc) has revolutionized the field of stem cell biology.66 In 2007, this technology was applied to human cells and it is now possible for investigators to generate induced pluripotent stem cells (iPSCs) from their patient's own skin biopsy-derived fibroblast cells using either the same cocktail of transcription factors or an independently determined mixture.67,68 The demonstration that human-derived iPSCs could be differentiated into functional cardiomyocytes was first reported by Zhang et al. in 2009.69
Subsequently, several channelopathy- and cardiomyopathy-specific, iPSC-derived cardiocyte models have been generated. For example, at least 5 different iPSC lines derived from LQTS patients have been published and in all of these studies, the investigators recapitulated the cardiac phenotype of prolonged repolarization and increased arrhythmogenesis associated with the disease.70–74 Additional studies have used iPSCs derived from LQTS patients to investigate new drugs for the treatment of the disease.74,75 The use of IPSCs has also been proposed to investigate the prolongation of action potentials due to potential drug-induced cardiotoxicity of novel and existing drugs in order to improve drug safety and drug development.76 This technology will allow more precise characterization of variants identified, in patients, as either pathogenic or benign, as well as examination of mechanisms underlying the reduced penetrance and variable expressivity that is common among cardiac electrical disorders by generating iPSCs from multiple related family members that show variable disease expression. The great anticipation is that iPSCs will facilitate the development of novel therapeutics as well as personalized treatment for patients with inherited diseases.
CONCLUSIONSOver the last 20 years, we have witnessed genomic advances that have deepened our understanding and appreciation for the role of genetics in cardiovascular diseases. As we continue to discover novel disease genes and mechanisms, the number of available genetic tests will surely increase. For the four specific heritable channelopathies/cardiomyopathies detailed in this review, we are continuing to expand our research knowledge about the underlying disease mechanisms that will continue to allow for new clinical translation and even better interpretation of genetic test results, enhancing our bench-to-bedside practice. The implementation of genetic testing as a diagnostic/prognostic tool for some of the cardiac diseases has propelled the community of cardiologists to become well versed in the language of genomic medicine and knowledgeable interpreters of the genetic test result. As new technologies, such as whole exome/genome sequencing and iPSC-generated cardiomyocytes improve and become more affordable, we will have new tools to enhance our goal of personalized medicine to render wise decisions to families being evaluated for the presence or absence of one of these potentially lethal yet highly treatable genetic cardiac arrhythmia disorders.
CONFLICTS OF INTERESTDr. Ackerman is a consultant for Biotronik, Boston Scientific, Medtronic, St. Jude Medical Inc., and Transgenomic. Intellectual property derived from MJA's research program resulted in license agreements in 2004 between Mayo Clinic Health Solutions (formerly Mayo Medical Ventures) and PGxHealth (formerly Genaissance Pharmaceuticals, now recently acquired by Transgenomic). There are no additional conflicts of interest.