Desminopathies are a largely autosomal dominant group of rare diseases caused by mutations in the desmin gene. Because desmin is the main component of intermediate filaments in cardiac, skeletal, and smooth muscle and of Purkinje fibers, these conditions are characterized by skeletal myopathy and cardiomyopathy (mainly restrictive) with arrhythmias or conduction disorders.1–3
The aim of our present study was to analyze the genotype and phenotype of patients with desmin mutation-related cardiomyopathy. Because published series normally include few patients, any further contributions will boost our understanding of this disease.
In 2 centers with 819 studied families, we analyzed all individuals found to have a desmin mutation after a phenotype-guided genetic study (restrictive cardiomyopathy/dilated cardiomyopathy with a restrictive pattern, families with high rates of pacemaker implants, skeletal myopathy, and/or creatine kinase elevation). Gene sequencing was performed using Sanger or next-generation sequencing. A pathogenic mutation was defined as any mutation involving an amino acid change from the reference sequence that met 3 criteria: it segregated with affected members of the family, it was not present in 200 chromosomes of healthy unrelated individuals, and the affected residue was conserved among species and desmin isoforms.
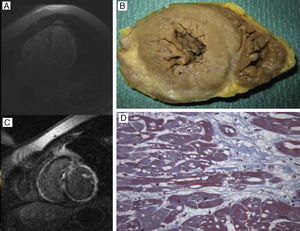
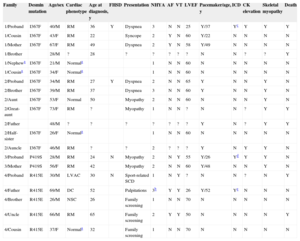
We studied 20 patients from 4 families, identifying 3 pathogenic mutations: Ile367Phe (2 families, Sanger), Pro419Ser (Sanger), and Arg415Gln (next-generation sequencing). Of these, 16 had desminopathy (including 2 obligate carriers who died of cardiomyopathy, without genetic confirmation) and 4 were young unaffected carriers (Table). The mean age at diagnosis was 35±15 years. Two families reported several relatives with pacemakers who died of cardiac arrest in their third or fourth decade of life. The most common symptoms were heart failure with an atrioventricular conduction disorder. The most common echocardiographic pattern was restrictive cardiomyopathy, with normal myocardial thickness or mild hypertrophy (11.4±2.4mm) and preserved left ventricular (LV) systolic function, except for 2 patients with severe LV dysfunction and another with LV arrhythmogenic cardiomyopathy (Figure). Cardiac magnetic resonance imaging was performed in 4 patients, who all had extensive transmural fibrosis; 7 of 11 men and 3 of 5 women required a pacemaker at early ages due to atrioventricular block, including 2 men who required ventricular resynchronization and 3 who required an implantable cardioverter-defibrillator due to nonsustained ventricular tachycardia.
Clinical and Genetic Data of the Families and Patients Included in this Study
| Family | Desmin mutation | Age/sex | Cardiac phenotype | Age at diagnosis, y | FHSD | Presentation | NHYA | AF | VT | LVEF | Pacemaker/age, y | ICD | CK elevation | Skeletal myopathy | Death |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/Proband | I367F | 40/M | RM | 36 | Y | Dyspnea | 3 | N | N | 25 | Y/37 | Yc | Y | Y | Y |
| 1/Cousin | I367F | 43/F | RM | 22 | Syncope | 2 | Y | N | 60 | Y/22 | N | N | N | N | |
| 1/Mother | I367F | 67/F | RM | 49 | Dyspnea | 2 | Y | N | 58 | Y/49 | N | N | N | N | |
| 1/Brother | 28/M | ? | 28 | ? | ? | ? | ? | ? | N | N | ? | ? | Y | ||
| 1/Nephewa | I367F | 21/M | Normala | 1 | N | N | 60 | N | N | N | N | N | |||
| 1/Cousina | I367F | 34/F | Normala | 1 | N | N | 60 | N | N | N | N | N | |||
| 2/Proband | I367F | 34/M | RM | 27 | Y | Dyspnea | 2 | N | N | 65 | Y | N | N | Y | N |
| 2/Brother | I367F | 39/M | RM | 37 | Dyspnea | 3 | N | N | 60 | Y | N | N | Y | N | |
| 2/Aunt | I367F | 53/F | Normal | 50 | Myopathy | 2 | N | N | 60 | N | N | N | Y | N | |
| 2/Great-aunt | I367F | 73/F | RM | ? | Myopathy | 1 | N | N | ? | N | N | ? | Y | Y | |
| 2/Father | 48/M | ? | ? | ? | ? | ? | ? | ? | Y | N | ? | Y | Y | ||
| 2/Half-sister | I367F | 26/F | Normala | 1 | N | N | 60 | N | N | N | N | N | |||
| 2/Auncle | I367F | 46/M | RM | ? | ? | 2 | ? | ? | ? | Y | N | Y | Y | N | |
| 3/Proband | P419S | 28/M | RM | 24 | N | Myopathy | 2 | N | Y | 55 | Y/26 | Yd | Y | Y | N |
| 3/Mother | P419S | 56/F | RM | 42 | Myopathy | 2 | N | N | 60 | Y/48 | N | N | Y | N | |
| 4/Proband | R415E | 30/M | LVAC | 30 | N | Sport-related SCD | 1 | N | Y | ? | N | N | ? | N | Y |
| 4/Father | R415E | 69/M | DC | 52 | Palpitations | 3b | Y | Y | 26 | Y/52 | Ye | N | N | N | |
| 4/Brother | R415E | 26/M | NSC | 26 | Family screening | 1 | N | N | 70 | N | N | N | N | N | |
| 4/Uncle | R415E | 66/M | RM | 65 | Family screening | 2 | Y | Y | 50 | N | N | N | N | Y | |
| 4/Cousin | R415E | 37/F | Normala | 32 | Family screening | 1 | N | N | 70 | N | N | N | N | N |
AF, atrial fibrillation; CK, creatine kinase; DC, dilated cardiomyopathy; F, female; FHSD, family history of sudden death; I367F, Ile367Phe; ICD, implantable cardioverter-defibrillator; LVAC, left ventricular arrhythmogenic cardiomyopathy; LVEF, left ventricular ejection fraction; M, male; MR, restrictive cardiomyopathy; NHYA, New York Heart Association classification of dyspnea; NSC, nonspecific cardiomyopathy; NSVT, nonsustained ventricular tachycardia; P419S, Pro419Ser; R415E, Arg415Glu; SCD, sudden cardiac death; VT, ventricular tachycardia.
 who shows biatrial and biventricular dilatation and extensive transmural fibrosis in the free wall of the left ventricle, septum, and right ventricle. B and D: Necropsy of the proband of family 4 (DES Arg415Glu mutation), revealing normal myocardial thicknesses and diameters, fiber hypertrophy, and severe subepicardial fibrosis, cytoplasmic vacuolization, and myofibrillar loss, which are all compatible with left ventricular arrhythmogenic cardiomyopathy.")
A and C: Cardiac magnetic resonance imaging of the proband of family 1 (DES Ile367Phe mutation) who shows biatrial and biventricular dilatation and extensive transmural fibrosis in the free wall of the left ventricle, septum, and right ventricle. B and D: Necropsy of the proband of family 4 (DES Arg415Glu mutation), revealing normal myocardial thicknesses and diameters, fiber hypertrophy, and severe subepicardial fibrosis, cytoplasmic vacuolization, and myofibrillar loss, which are all compatible with left ventricular arrhythmogenic cardiomyopathy.
The degree of skeletal myopathy was variable and mainly involved distal and progressive muscle weakness and atrophy of the lower limbs. Penetrance was high from the third decade of life, with more severe expression in men.
Six patients died during follow-up: 1 from cardiac arrest (a 30-year-old man with LV arrhythmogenic cardiomyopathy), 2 from heart failure (a 40-year-old man with severe LV dysfunction and a 66-year-old man with restrictive cardiomyopathy), and 3 from unknown causes (1 had a pacemaker).
Few data are available on these 3 mutations because they have only been described in the literature in 4 families: 1 family with Ile367Phe,1,2 3 with Pro419Ser,1,2,4 and none with Arg415Glu.
The Ile367Phe mutation has previously been described as being pathogenic.2 Segregation of the mutation with the disease was checked in our 2 families and the phenotype was similar to that already described: restrictive cardiomyopathy, atrioventricular block, and skeletal myopathy.
The Pro419Ser mutation has also been described and shows a more variable phenotype: marked neurological involvement with predominantly distal muscle weakness and a nasal voice, restrictive cardiomyopathy, and atrioventricular block and, in the other family, right ventricular arrhythmogenic cardiomyopathy.2,4 This mutation is located in the tail of the gene, and cardiomyopathy typically appears alone or precedes a skeletal condition. In contrast, the Ile367Phe mutation is located in a hot spot and is the most severe mutation.2
The Arg415Glu mutation in exon 6 has not been described before and affects splicing. Cosegregation was seen in our family. The proband died of sudden cardiac death, with histopathology findings of LV arrhythmogenic cardiomyopathy, and is the first published patient with this association. This mutation was seen in most of our patients with severe cardiomyopathy without skeletal myopathy.
Notably, these patients visited the clinic with other diagnoses, such as dilated hypertensive heart disease, or hypertrophic cardiomyopathy. The fourth proband was diagnosed after sudden cardiac death. The cause was suggested by a specialized study and confirmed by genetic analysis. A subsequent family study diagnosed 14 carriers, 10 with restrictive cardiomyopathy and/or skeletal myopathy and 4 unaffected young carriers. The study also allowed 16 families to be excluded from follow-up.
In conclusion, desminopathies commonly present as restrictive cardiomyopathy with heart failure in the third or fourth decade of life or early advanced atrioventricular block requiring pacemaker implantation and, in some patients, additionally, implantable cardioverter-defibrillator due to nonsustained ventricular tachycardia.
FUNDINGRed Investigación Cardiovascular (RIC) of the Instituto de Salud Carlos III (PI14/01477, RD12/0042/0029, RD12/0042/0069), FEDER and CIBEROBN (CB12/03/30038), Madrid, Spain.