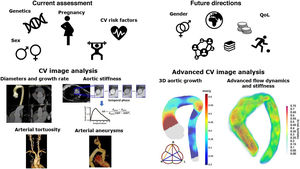
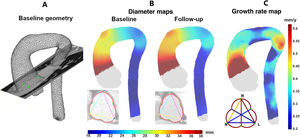
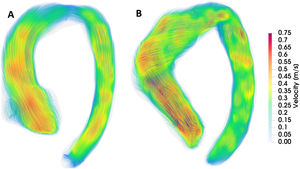

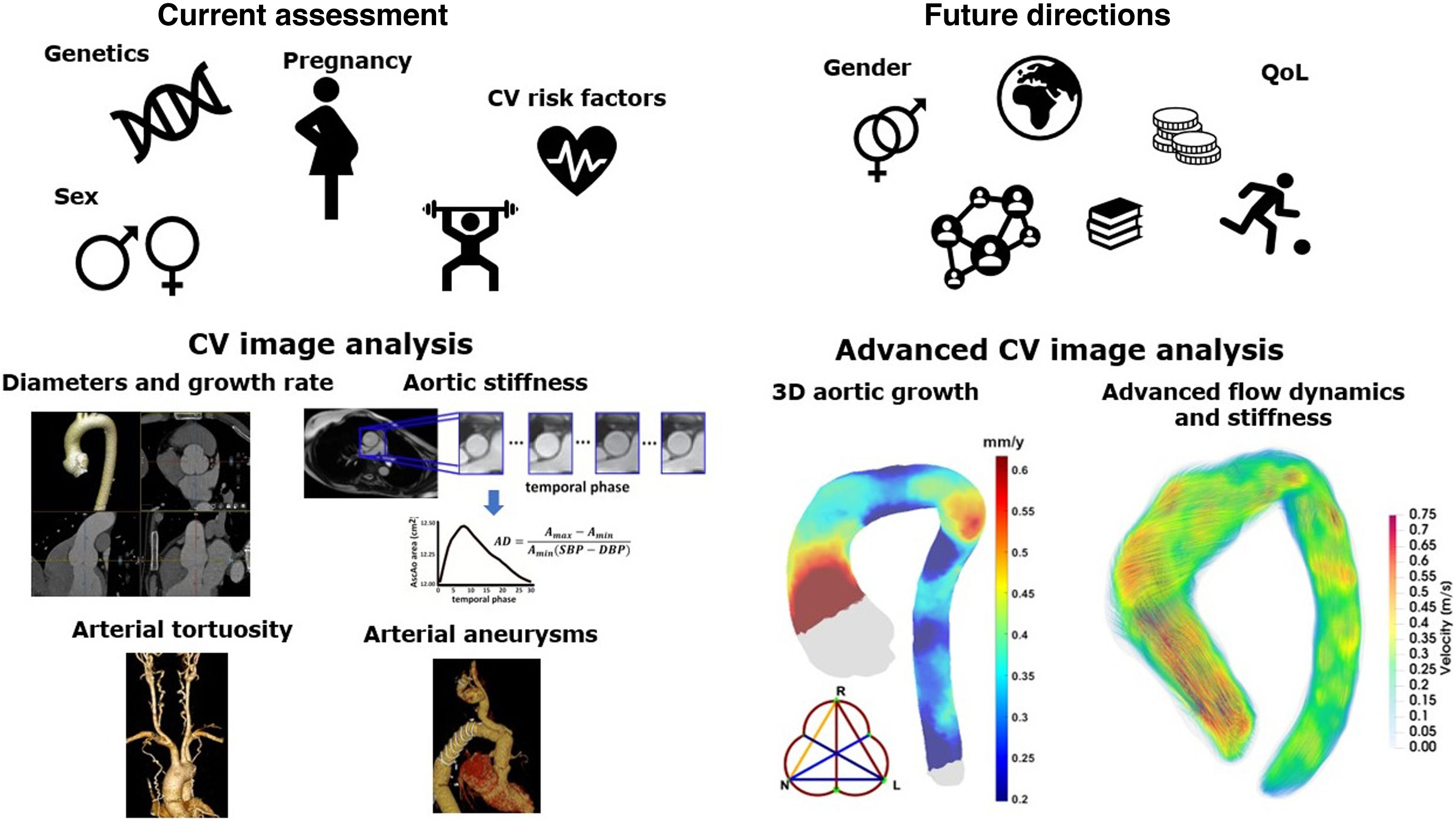
Heritable thoracic aortic diseases (HTAD) are a group of diverse genetic conditions characterized by an increased risk of aortic complications. The standard surveillance of these patients involves monitoring aortic diameters until a defined threshold is reached, at which point preventive aortic surgery is recommended. However, assessing aortic risk in these patients is far more complex and, in many aspects, remains incompletely understood. Several factors contribute to this complexity, including the diversity and low prevalence of the conditions within HTAD and the limited understanding of the factors influencing the progression of aortic dilation and the advent of acute aortic events. This article reviews current knowledge on clinical, genetic, and imaging factors related to aortic risk in HTAD and explores their potential future roles in improving risk assessment. By advancing our understanding of these factors, we aim to enhance the precision of risk stratification and develop more effective, personalized management strategies for HTAD patients, with the final goal of improving clinical outcomes and quality of life in individuals affected by these genetic disorders.
Keywords
Identify yourself
Not yet a subscriber to the journal?
")
Purchase access to the article
By purchasing the article, the PDF of the same can be downloaded
Price: 19,34 €
Phone for incidents
Monday to Friday from 9am to 6pm (GMT+1) except for the months of July and August, which will be from 9am to 3pm