Even when reperfusion therapy is applied as early as possible, survival and quality of life are compromised in a considerable number of patients with ST-segment elevation acute myocardial infarction. Some cell death following transient coronary occlusion occurs during reperfusion, due to poor handling of calcium in the sarcoplasmic reticulum-mitochondria system, calpain activation, oxidative stress, and mitochondrial failure, all promoted by rapid normalization of intracellular pH. Various clinical trials have shown that infarct size can be limited by nonpharmacological strategies—such as ischemic postconditioning and remote ischemic conditioning—or by drugs—such as cyclosporine, insulin, glucagon-like peptide-1 agonists, beta-blockers, or stimulation of cyclic guanosine monophosphate synthesis. However, some clinical studies have yielded negative results, largely due to a lack of consistent preclinical data or a poor design, especially delayed administration. Large-scale clinical trials are therefore necessary, particularly those with primary clinical variables and combined therapies that consider age, sex, and comorbidities, to convert protection against reperfusion injury into a standard treatment for patients with ST-segment elevation acute myocardial infarction.
Keywords
Ischemic heart disease is the leading cause of death worldwide and, unless previous trends are modified, will continue to be the most common cause of death in 2030.1 The social impact of ischemic heart disease is considerable, not only because of the mortality it causes, but also because of its consequent morbidity, loss of quality of life, and high economic cost. This impact is largely due to a pathophysiological mechanism, namely, cardiomyocyte death. In ischemic heart disease, cardiomyocyte death almost always occurs in the context of severe and prolonged myocardial ischemic events, which are a consequence of thrombotic complications from atherosclerotic plaques in epicardial coronary arteries, known as acute coronary syndrome. Cardiomyocyte death is more significant when ischemia is caused by complete coronary occlusion, lacks well-developed collateral circulation, affects the majority of the left ventricular wall thickness, and shows ST-segment elevation on an electrocardiogram (ST-segment elevation acute myocardial infarction [STEMI]).
The cell death that occurs in acute coronary syndrome not only causes a direct loss of contractile activity, but can also cause geometric changes in the infarcted wall and adaptive changes in the remaining myocardium, which ultimately lead to general dysfunction and dilatation of the ventricle, a process called adverse remodeling.2,3 Scarring and adverse remodeling cause heart failure and promote the appearance of potentially fatal ventricular arrhythmias,4 so that cell death occurring during acute coronary syndrome eventually determines not only acute-phase mortality, but also long-time morbidity and mortality.5
Accordingly, reducing cell death during acute coronary syndrome, and in particular during STEMI, appears to be an obvious strategy for reducing the impact of ischemic heart disease on health and society.
Prevention of Reperfusion Injury as a Strategy to Reduce the Impact of Ischemic Heart DiseaseIschemic heart disease has mixed genetic and environmental etiology whose pathological substrate is the atherosclerotic plaque. Coronary atherosclerosis develops in a clinically silent manner over years and only causes clinical symptoms when the vessel lumen is greatly narrowed, either due to atherosclerotic plaque growth or the development of intracoronary thrombosis due to plaque complication, resulting in a loss of endothelial continuity due to erosion, fissure, or endothelial rupture.6 Thus, preventing the appearance of atherosclerosis coronary plaques or their progression is the first line of action against the disease.
Prevention of ischemic heart disease is not without difficulties. Population-wide interventions are expensive and cannot be particularly aggressive due to the risk of costly adverse events. The more aggressive interventions—aimed at avoiding plaque growth, reducing the risk of complications, or attenuating secondary thrombosis—should be limited to those individuals at high risk of the disease. Although much progress has been made in identifying risk factors and developing methods to calculate individual risk,7 conventional risk factors for the disease are poorly controlled.8 Moreover, genetic studies have identified multiple loci associated with the development of atherosclerosis disease, but this association is weak and its effect on the predictive value of conventional risk factors is limited, particularly when family history is taken into account. The difficulty in preventing ischemic heart disease is clear upon observing changes in its incidence in specific populations. Spain, for example, is far from controlling ischemic heart disease through preventive measures, given that the incidence of conventional risk factors has largely been stable and in some cases is predicted to increase, as is happening with type 2 diabetes mellitus, which has proliferated due to the growth in childhood obesity.9,10
The effectiveness of risk stratification and prevention strategies should be measured not only by their ability to reduce the appearance of clinical symptoms of ischemic heart disease, but more specifically by their ability to reduce the incidence of acute coronary syndrome and, in particular, acute myocardial infarction (AMI), which causes most of the morbidity and mortality associated with the disease. However, extensive evidence indicates that it is difficult to predict the occurrence of AMI. In a recent large study, performed in 542 008 patients with a first myocardial infarction, approximately half had only one risk factor or none at all.11,12 Moreover, analysis of distinct patient subgroups revealed that in-hospital mortality was inversely proportional to the number of classical risk factors present.11 Taking both observations together, it can be estimated that most patients that die in hospital from their first AMI show a low risk profile, with 0-1 risk factors. Accordingly, although efforts to improve risk stratification and prevention of ischemic heart disease are clearly essential, no less essential is the need to improve the effectiveness of AMI treatment to improve prognosis. In many cases, this approach is the first and only option available to alter the course of the disease.
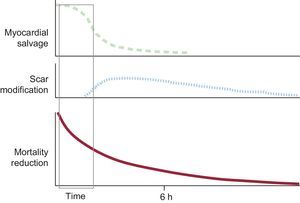
The prognosis of AMI largely depends on its extent, that is, the number of cells that die during the event.13 The final necrotic extent is mostly due to the speed of the progression of ischemic injury (influenced by residual blood flow, whether through the lesion or through collateral circulation, and by temperature, among other factors) and the duration of the ischemia.14,15 The most effective treatment to limit infarct size is early reperfusion.16–18 However, the amount of myocardium salvaged by reperfusion rapidly decreases the longer it is delayed, and the window in which reperfusion effectively limits infarct size in patients with STEMI is short. After 3h of ischemia, without collateral circulation and residual flow (TIMI [Thrombolysis in Myocardial Infarction] 0), the amount of salvaged myocardium is typically small or nonexistent.19 Nonetheless, late reperfusion is also beneficial, and should generally be performed within 12h of symptom onset.20 In this case, however, the benefit is due to the positive effects of reperfusion on healing, with reperfusion limiting infarct expansion and secondary adverse remodeling (Figure 1).20

Because of the short reperfusion window for limiting infarct size, necrosis affecting most of the ventricular wall thickness-causing Q-waves and worsening prognosis-cannot be prevented in most patients, despite the availability of systems of rapid administration of reperfusion treatment (such as primary angioplasty or thrombolysis without emergency department referral, and optimal antiplatet and anticoagulant agents, etc. The conventional outlook is that when patients with STEMI arrive at the catheterization laboratory, as it is no longer possible to reduce the ischemia duration or modify the collateral circulation or residual coronary flow (TIMI flow), the only remaining approach is to open the artery as much as possible and see if patients have arrived in time to save the myocardium. However, a wealth of preclinical information and a growing amount of clinical data indicate that the proportion of reperfusion-salvaged myocardium can be increased by applying treatments more or less at the time of reperfusion. The present article examines the current situation and the expected future of treatments aimed at reducing cell death in patients with STEMI that receive reperfusion treatment, and will not therefore analyze the effectiveness of these treatments in other clinical contexts, such as cardiac surgery or cardiopulmonary resuscitation.
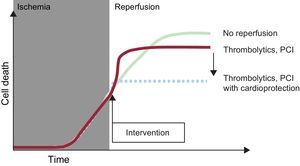
REPERFUSION INJURYConceptThe first laboratory experiments with transient coronary occlusion showed a paradoxical worsening during reperfusion of the functional changes associated with ischemia, particularly arrhythmias.21–23 Studies in isolated and perfused hearts revealed an increase in cardiac enzyme release upon reperfusion following a period of ischemia.24 Afterward, it was verified that reperfusion was accompanied by transient contractile dysfunction (stunning), even in the absence of necrosis.25 However, differences in the mechanisms and clinical relevance of these reperfusion-associated phenomena hampered initial acceptance of the concept of reperfusion injury in the scientific and medical community. The situation changed following the introduction of the concept of lethal reperfusion injury, with a precise operational definition: cell death that can be prevented by interventions applied at the time of reperfusion.26 In this article, the expression “reperfusion injury” is always used according to this definition, whose use for STEMI is illustrated in Figure 2.27

Illustration of the concept of reperfusion injury. During myocardial ischemia, cell death progresses. Reperfusion halts this process, but its benefit is limited by an increase in cell death during the first few minutes of reperfusion; this death can be avoided by using cardioprotective treatments at the time of flow restoration. PCI, percutaneous coronary intervention. Modified with the permission of Garcia-Dorado et al.27.
Occlusion of a coronary artery creates an ischemic zone (area at risk) whose lateral borders are sharply defined by a lack of communication between adjacent capillary beds28,29 and in which residual blood flow, if any, is distributed through the subepicardium. Reperfusion is accompanied by extracellular and intracellular edema, due to endothelial permeability and the creation of an osmotic gradient between the extravascular and intravascular spaces.30 The extracellular edema appears a few minutes after reperfusion and can contribute to changes in the mechanical function of the myocardium and last for several days in patients with STEMI, which is how it has been used to delimit the area at risk though magnetic resonance imaging.30 In addition to edema, reperfusion is accompanied by the accumulation of platelets in the reperfused myocardium due to their adhesion (dependent on P-selectin) to activated endothelium.
An area of cell death is produced if reperfusion is not performed sufficiently early. Cell death rapidly occurs due to necrosis in the first few minutes of reperfusion.26 In contrast to what was once thought, apoptosis plays no significant role in ischemia-reperfusion–induced cell death; in fact, the expression of proteins indispensable for the apoptotic program is silenced in adult cardiomyocytes.31 The necrotic zone has a lateral extent that coincides with the lateral extension of the area at risk, and a transmural extent that extends toward the subepicardium as reperfusion is delayed (wavefront phenomenon).14 When reperfusion is late and the infarction is extensive, the interior develops areas of severe microvascular injury with loss of the endothelial barrier, interstitial bleeding, and flow arrest (no-reflow areas).32 No-flow areas are associated with extensive infarctions and poor prognosis, suggesting that they contribute to cell death. However, there is no definitive evidence to support this theory, and considerable data indicate that no-flow areas are produced in already necrotic zones.33 What does seem likely is that areas without reperfusion hamper healing and favor scar expansion and subsequent adverse remodeling.34
Ischemic Cell DamageReperfusion injury, as defined above, is not limited to STEMI, in which cell death is produced secondary to transient myocardial ischemia and can occur in diverse clinical situations, particularly during cardiac surgery or cardiopulmonary resuscitation. However, the mechanisms involved depend on the changes produced before reperfusion (such as concomitance of hypothermia, cardioplegic solutions, hypoxia with preserved blood flow, etc. This article focuses on the mechanisms of reperfusion injury that appear after a period of severe regional and normothermic ischemia.
The cell changes caused by ischemia have been reviewed in numerous publications.26,35–38 Within seconds, regional ischemia causes oxidative phosphorylation arrest due to a lack of oxygen in the mitochondrial respiratory chain. Adenosine triphosphate (ATP) synthesis becomes restricted to the glucolytic pathway, which, with the Krebs cycle stopped, leads to an intracellular accumulation of lactic acid.26,38 This buildup of lactic acid, together with the inability to eliminate CO2 from the extracellular space due to blood flow arrest, causes intracellular pH to rapidly fall to almost 6.4 within minutes.39,40 Acidosis and accumulation of inorganic phosphate rapidly halt contractile activity, even when the ATP concentration remains practically normal. In the following minutes, the ATP concentration continues to fall, reaching critical and almost zero levels and promoting the development of ischemic rigor.41 Depletion of ATP is accelerated in the final phase by a reversal in the activity of mitochondrial ATPase (complex V of the respiratory chain), which consumes ATP to maintain the mitochondrial membrane potential.42 Acidosis activates sarcolemmal Na+/H+ exchangers, with a consequent influx of Na+, while the energy depletion stops Na+ efflux through Na+/K+ ATPase. Consequently, Na+ accumulates in the interior of the cell, activating Na+/Ca2+ exchangers in the reverse direction and increasing cytosolic Ca2+.26,38 The cell becomes depolarized. At the initial stage of ischemia, mitochondria take up Ca2+ due to the electronegativity of the mitochondrial matrix, but cease to do so when depolarized.43 The sarcoplasmic reticulum cannot take up Ca2+ from the cytosol either, as the transporter needs ATP to work.
Reperfusion InjuryRestoration of myocardial blood flow leads to a recovery in respiratory activity, mitochondrial membrane potential, and ATP synthesis. The cell membrane potential is restored, which, in the presence of a high cytosolic concentration of Na+, causes the Na+/Ca2+ exchanger to function in reverse, further exacerbating the cytosolic Ca2+ overload. The availability of ATP in the presence of increased Ca2+ activates uptake of Ca2+ by the sarcoplasmic reticulum, exceeding the threshold necessary to open ryanodine channels, which release Ca2+ into the cytosol. Cyclical repetition of this process causes Ca2+ oscillations that are propagated through the cell.26,38 Ryanodine channels are physically connected to mitochondria, so that Ca2+ is specifically released into subcellular microdomains, allowing influx along the potential gradient into the mitochondrial matrix via the mitochondrial Ca2+ uniporter. The physicochemical characteristics of this transporter cause the Ca2+ oscillations to promote an increase in mitochondrial Ca2+. Ca2+ oscillations and their effect on mitochondria depend, on one hand, on the activity of the ryanodine channels and the Ca2+ uniporter and, on the other, on the activity of the responsible transporter, which in turn is modulated by phospholamban.44–46 During reperfusion, activation of protein kinase A, protein kinase G (PKG), and calcium- and calmodulin-dependent oxidation/phosphorylation of kinases modulate the magnitude of the Ca2+ oscillations and the degree of mitochondrial Ca2+ overload, through changes in the phosphorylation of ryanodine, phospholamban, and mitochondrial Ca2+ uniporter channels.47
Cytosolic Ca2+ oscillations have detrimental consequences for cells. First, the oscillations promote an excessive contractile activity, which can trigger hypercontracture.48 Hypercontracture can disrupt cellular architecture and cause sarcolemmal rupture and cell death.49 Second, the Ca2+ oscillations lead to cell membrane potential oscillations in the form of early depolarizations or afterdepolarizations, an important mechanism for reperfusion arrhythmias.50 Third, the oscillations promote opening of the mitochondrial permeability transition pore (mPTP).
The mPTP is a high-permeability channel, of poorly understand molecular identity, that permits a direct connection between the mitochondrial matrix and intermembrane space.51 When the channel opens, the mitochondrial membrane potential dissipates and ATP synthesis stops, resulting in edema and mitochondrial rupture and allowing the release of mitochondrial molecules to the cytosol, including Ca2+. Mitochondrial permeabilization is considered to be an important mechanism of cell death during reperfusion. Permeabilization and hypercontracture secondary to Ca2+ overload and oscillations are interrelated, so that oscillations favor opening of the mPTP52 and mitochondrial permeabilization promotes Ca2+ efflux into the cytosol, which promote further development of oscillations and hypercontracture.53 Hypercontracture appears to play a more important role in the genesis of cell death after relatively brief ischemic events, whereas mPTP is the main mechanism of cell death during reperfusion after prolonged ischemia.54
Both hypercontracture and mPTP opening seem to be favored by 2 important phenomena that occur during the first few minutes of reperfusion. The first is the rapid normalization of intracellular pH, which permits contractile activity and mPTP opening, both inhibited by acidosis.55,56 pH normalization also allows activation of calpains, Ca2+-dependent proteases that are inhibited by an acidic pH. Calpain activation damages the cytoskeleton and impedes the normal function of Na+/K+ ATPases, which worsens the Na+ and Ca2+ overload and closes the vicious circle.57–59
During reperfusion, reactive oxygen species are generated that play a crucial role in reperfusion injury, because they directly promote opening of the mPTP60 and worsen changes in Ca2+ homeostasis, oxidizing calcium-dependent kinase and calmodulin.61 Moreover, superoxide anion, which is a potent oxidant, dissociates nitric oxide (NO) synthase from tetrahydrobiopterin, resulting in superoxide anion production instead of NO and thereby decreasing the levels of NO and its downstream signaling pathways (cyclic guanosine monophosphate [cGMP] and PKG).62 The reduction in PKG accelerates pH normalization (as PKG inhibits Na+/H+ exchange) and promotes Ca2+ oscillations (acting on phospholamban); as explained above, both actions have negative consequences for cell survival.
Finally, cell death can be propagated between adjacent myocytes through gap junction-mediated intercellular communication,63–66 which allows passage of Na+ from one cell to another.67 Na+, in turn, promotes Ca2+ influx into adjoining cells, which can cause death if the cells are sufficiently damaged (eg, if the Na pump function is altered or the cytoskeleton is damaged).67 Intercellular communication through gap junctions also requires pH normalization, given that acidosis causes these channels to close.68
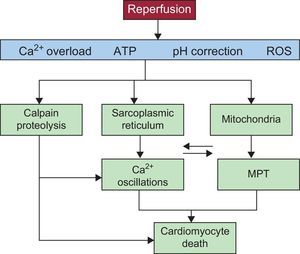
These mechanisms are summarized in Figure 3.
 (see the detailed explanation in the text). ATP, adenosine triphosphate; MPT, mitochondrial permeability transition; ROS, reactive oxygen species.")
Diagram showing the main mechanisms of cardiomyocyte death during myocardial reperfusion. Reperfusion causes calcium oscillations that are dependent on the sarcoplasmic reticulum and mitochondrial permeabilization that is promoted by calcium overload and oxidative stress. Recovery from acidosis promotes both phenomena, which are intimately related due to physical contact between mitochondria and sarcoplasmic reticula and also the activation of calcium-dependent proteases (calpain) (see the detailed explanation in the text). ATP, adenosine triphosphate; MPT, mitochondrial permeability transition; ROS, reactive oxygen species.
Both myocardial ischemia and reperfusion actually occur in a tissue containing a considerable number of nonmyocyte cells, such as endothelial, fibroblastic, and blood cells. Activated platelets adhering to the reperfused myocardium negatively affect cardio-myocytes, by mechanisms independent of microvascular flow occlusion and probably related to the release of substances such as thrombin. Thus, activated platelets can worsen Ca2+ management and trigger other adverse events in cells.69–72 Endothelial cell activation and microparticle and ribosomic RNA release also impair myocyte survival via massive activation of tumor necrosis factor alpha, among other factors.73
THE MOST PROMISING STRATEGIES FOR LIMITING REPERFUSION INJURYDemonstration of the existence of reperfusion injury used to be based on the use of pharmacological tools that interfered with the above described mechanisms. However, for reasons discussed below, none of these tools has been translated to clinical practice. Thus, drugs remain to be developed for many potentially useful targets for treating reperfusion injury.74
Endogenous CardioprotectionThe first treatment successfully used in patients with STEMI was ischemic postconditioning (PostC).75,76 In contrast with drug treatments, PostC reduces reperfusion injury by introducing brief episodes of ischemia, at the moment when reperfusion begins,77 which has a protective effect without the need for external agents (endogenous cardioprotection).
PostC acts via mechanisms different from ischemic preconditioning, in which brief and repeated periods of ischemia immediately before a longer episode reduce the resulting cell death. Ischemic preconditioning provides a more robust protection, but is unsuitable for patients with STEMI. The protection afforded by PostC is not only lesser, but varies depending on conditions. Its effectiveness is weak or absent after brief ischemic episodes that cause small infarcts.78 The protective effect of PostC is mainly due to its ability to delay the normalization of intracellular pH for a few minutes, by slowing metabolite washout secondary to flow interruptions, and to the reduction in oxidative damage, which preserves the NO-cGMP-PKG signaling pathway and inhibits Na+/H+ exchange.79,80
Recently, a form of endogenous protection has been described that involves remote induction (generally in the extremities) of intermittent myocardial ischemia immediately before or in the first few minutes of reperfusion: remote ischemic conditioning (RIC).81–83 One advantage of RIC over PostC is that it does not require manipulation of the coronary artery during the first few minutes of reperfusion, but the mechanisms by which it exerts its protective effect remain unclear.83,84
Pharmacological CardioprotectionPreservation of the PKG signaling pathway has many protective effects. As well as delaying pH normalization and reducing Ca2+ oscillations, as described above, the PKG pathway seems to have direct mitochondrial effects that inhibit mitochondrial permeabilization.85 In fact, there is ample evidence of the protective effects of treatments that normalize cGMP at the beginning of reperfusion, either through stimulation of particulate guanylate cyclase via natriuretic peptides86 or through direct and NO-independent stimulation of soluble guanylate cyclase.87 The usefulness of the stimulation of soluble guanylate cyclase via exogenous NO varies, probably because excess NO can cause oxidative damage.88
Another strategy that has been widely studied using both pharmacological interventions89,90 and genetically modified models90 involves inhibition of mPTP opening. Both investigational approaches focus on one of the proteins that participates in Ca2+-induced mPTP opening, cyclophilin D. Cyclosporine, a widely used immunosuppressor, acts through cyclophilin D to inhibit mPTP opening89,90 in response to Ca2+. Preliminary results are available on other molecules that inhibit the mPTP. More recently, dysfunction of complex I of the respiratory chain has been identified as a potentially useful target. Although various ways to prevent this dysfunction have been proposed, there are few results to date.
Preclinical studies are being undertaken involving other targets. The most prominent targets are related to insulin signaling and energy metabolism. For the former, the early use of solutions containing glucose, insulin, and potassium91-93 and stable mimetics of glucagon-like peptide-194 have received particular attention and have achieved promising results. Other potentially protective ways of altering energy metabolism at the time of reperfusion include use of trimetazidine95 and the addition of specific substrates.96
Finally, there are many other interventions directed at distinct reperfusion injury targets. However, their effects remain controversial. These interventions include targeting activation of leukocytes or complement factors (which occurs when most cell death has taken place) or caspase-dependent apoptosis (a type of cell death practically inexistent in adult cardiomyocytes).
CLINICAL TRIALSDuring the last decade, various clinical trials have tested different strategies for limiting infarct size and reducing reperfusion injury. Their target populations, sample sizes, designs, primary end points, and results have recently been re-viewed.97-100 All of the studies lacked statistical power to detect effects on clinical course and were designed to detect effects on surrogate markers, specifically infarct size, measured or estimated by distinct methods. Some strategies, such as PostC, have been the subject of multiple studies, including meta-analyses, whereas only data from single studies are available for other interventions. Next, the results obtained with distinct cardioprotective interventions are assessed.
Ischemic ConditioningIn general, studies based on ischemic conditioning maneuvers have been positive.101
Ischemic PostconditioningSeveral studies have analyzed the efficacy of ischemic PostC, the first approach described as being able to limit reperfusion injury in patients. In the first trial, reocclusions were performed in the reperfused artery through four 1-min inflations of an angioplasty balloon, separated by 1-min periods with the balloon deflated and beginning within 1min of artery opening. This protocol significantly reduced infarct size, measured as the area under the curve for creatine kinase.75 Subsequent studies performed by other groups obtained varying results,76,102–105 whose differences may be explained by the involvement of distinct factors, such as different levels of direct stent usage or differences in perioperative antiplatelet therapy.103 Although the current data generally show that PostC reduces infarct size to a certain extent,106 the use of this procedure has not appreciably expanded in recent years, probably due to the weak and variable protection that it appears to provide, its counterintuitive character (producing reocclusions), and fear of complications due to the repeated inflations of the angioplasty balloon. One limitation common to the different clinical assays of PostC is that this procedure cannot be performed in a double-blind manner. This is an important limitation that may introduce biases in patient exclusion, event allocation, and incompletely automated determinations, such as in the case of infarct size evaluation via magnetic resonance imaging.107
Remote Ischemic ConditioningAs with PostC, RIC was applied to patients soon after being described in laboratory studies and long before the mechanisms were understood.108 RIC can be induced by inflating a blood pressure cuff placed on the arm to above systolic pressure for 5-min periods separated by 5-min intervals. The results described thus far are consistently positive and in agreement with the preclinical evidence. RIC has multiple advantages over PostC and is a strong candidate for becoming part of clinical practice recommendations. First, it is extremely safe and cheap; second, it can be applied in the ambulance and to all patients with STEMI that receive reperfusion treatment, not only those who undergo primary angioplasty; and, third, the protection afforded by RIC appears to go beyond the myocardial area at risk and STEMI. Studies are underway in other tissues,109 and there is already solid evidence that it reduces myocardial injury in various types of surgery,110,111 particularly coronary revascularization.112,113 Although it is complicated to perform double-blind studies of RIC, it is possible, as the placebo treatment could involve cuffs automatically programmed to inflate to a low pressure.114
Pharmacological TreatmentsThe results of pharmacological treatments to reduce infarct size in STEMI are as variable as the strategies and designs used. However, studies based on approaches with robust preclinical data and designs that faithfully reproduce the conditions of preclinical studies have generally been positive.108,115–119
Atrial Natriuretic PeptideThe cGMP/PKG signaling pathway is probably the pharmacological target for the prevention of myocardial reperfusion injury that has the most solid preclinical evidence. However, only 1 clinical trial has directly assessed this strategy in patients with STEMI.115 The study showed limitation of the enzymatic infarct size and improvement in ventricular function in patients that received intravenous atrial natriuretic peptide at the time of reperfusion, thereby reproducing the beneficial effect of this treatment in animals.86
Glucose-insulin-potassiumInsulin administration also acts through the cGMP/PKG pathway (via Akt and PI3P), as well as by increasing glucose use.120,121 Preclinical evidence showing the usefulness of insulin at the beginning of myocardial reperfusion is solid.91,120–123 However, translation to the clinical setting gave negative results, easily explained by late treatment administration.124–126 A recent randomized, double-blind study has found, however, that glucose-insulin-potassium can reduce infarct size and serious electrical complications when administered in the ambulance to patients with STEMI that will receive primary angioplasty.117
Glucagon-like Peptide-1Stimulation of the glucagon-like peptide-1 pathway also acts through cGMP/PKG, among other pathways.127,128 There is solid evidence of the cardioprotective effect of increasing glucagon-like peptide-1–dependent signaling, either by using stable analogues of glucagon-like peptide-1 or inhibitors of its degradation.129,129 Two clinical trials have evaluated the effect of a glucagon-like peptide-1 agonist, exenatide, in patients with STEMI who underwent primary angioplasty, and both studies have shown positive results.118,130 Given its high safety profile, this strategy is especially promising.
AdenosineAdenosine, a degradation product of adenosine monophosphate, has a potent vasodilatory action, among other effects. In addition to its effects on the vasculature and leukocytes, it directly acts on cardiomyocytes, increasing NO availability (via Akt) and, therefore, activating the cGMP/PKG pathway. This substance was used intravenously in the AMISTAD studies,131 without conclusive results, although with evidence of protection at high doses in patients with previous infarctions.132 Subsequently, its use was proposed in the form of an intracoronary injection before reperfusion.133 The largest study performed to date is PROMISE, a double-blind study that included 200 patients with TIMI flow 0-1 and whose main end point was magnetic resonance imaging measurement of infarct size, with a prespecified subanalysis according to ischemia duration. The results showed that adenosine fails to limit infarct size in all patients, but did achieve this goal in those receiving primary angioplasty within the first 3h after the onset of pain.134 The data also suggest an improvement in left ventricular ejection fraction during the first 6 months after the infarction (shown by magnetic resonance imaging). Because intracoronary adenosine is a safe and inexpensive treatment, its early use in reperfused patients could be easily adopted if later studies, based on clinical end points, confirm the benefits.
CyclosporineIn a first pilot study, cyclosporine A limited infarct size (determined enzymatically and with magnetic resonance imaging) in 58 patients with STEMI undergoing primary angioplasty.119 However, a recent study with a similar number of patients found no benefit.135 A soon-to-be-completed international study with more than 1600 patients will presumably determine the usefulness of this treatment for improving the clinical course of patients with STEMI.136
Beta-blockersPreclinical evidence on the use of beta-blockers for reducing infarct size is mixed. A recent study found that intravenous injection of metoprolol was protective in a pig model involving a 90-minute coronary occlusion.137 This treatment was translated to patients in the METOCARD-CNIC study.138 This blinded study aimed to determine the effect of intravenous metoprolol, administered immediately before primary angioplasty, on infarct size determined with magnetic resonance imaging; the approach showed protection via an increase in myocardial salvage without major complications.138,139 Beta-blockers are already recommended for patients with STEMI. Therefore, the data from this study will advance the clinical use of beta-blockers, as long as they are confirmed by subsequent studies.
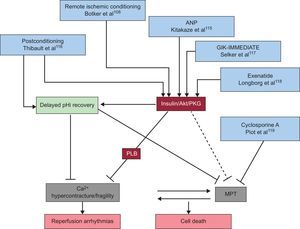
Some of the more promising strategies for combating reperfusion injury in patients with STEMI are shown in Figure 4.

Some of the more promising strategies for combating reperfusion injury, by the mechanism of cell death interrupted. ANP, atrial natriuretic peptide; GIK, glucose-insulin-potassium; MPT, mitochondrial permeability transition; pHi, intracellular pH; PKG, protein kinase G; PLB, phospholamban.
Cardioprotective therapy use in clinical practice has occasionally led to negative studies. In the overwhelming majority of cases, the negative results were clearly foreseeable from existing knowledge. The most common reasons for negative results are the following: a) use of treatments indicated by preclinical data to require administration before ischemia occurrence, such as Na+/H+ exchanger inhibitors, which have been shown to be effective only when they are administered before ischemia140-142 and failed when they were given at the time of reperfusion in clinical studies143; b) late treatment initiation, as multiple trials have begun study therapies after the first 30min of reperfusion, when most of the reperfusion injury has already occurred, as in the case of studies with glucose-insulin-potassium124 and erythropoietin,144 and c) therapeutic approaches with clearly insufficient or contradictory preclinical evidence, such as that of mechanical ventricular assist during reperfusion,145 protein kinase C-delta inhibition,146 complement system inhibition,147 or nicorandil.115
In addition to an inadequate poorly justified or risky design, other factors can hinder translation of experimental data to the clinical setting.100 One very important factor is advanced age and comorbidities, which contrasts with the use of healthy animals and young people in laboratory studies. Another factor is the simultaneous use of therapies that may have a protective effect or interfere with that of the treatments.97 Finally, the high variability in infarct size and residual flow or collateral circulation in patients with AMI hampers the detection of differences between groups.97,100 To avoid these problems, guidelines have been proposed for preclinical research and the design of clinical studies into protection against reperfusion injury.19
Finally, evidence that an intervention reduces infarct size in patients with STEMI is insufficient to justify its use in clinical practice. Studies must be performed with robust main clinical end points, which require a large number of patients with confirmed clinical benefit.
PROTECTION AGAINST REPERFUSION INJURY IN CLINICAL PRACTICE, NOW AND IN THE IMMEDIATE FUTUREAll the therapies against reperfusion damage in STEMI described so far are currently experimental. However, protection against myocardial damage can be discussed in certain clinical practice contexts. First, preclinical studies have demonstrated that some treatments received by patients with STEMI have a cardioprotective effect. These treatments include morphine and its derivatives,148 P2Y12 inhibitors and other antiplatelet agents,149 statins,150 and beta-blockers.137 Second, there are treatments that, due to their ease of use and safety, could be adopted before there is solid evidence of their real clinical usefulness. PostC is one such treatment. This treatment category could also encompass RIC.
Currently, studies are underway to evaluate treatments against reperfusion injury using surrogate markers, as well as other large studies that use clinical end points. The next frontier will probably have 2 fronts, involving, on one hand, personalization of antireperfusion therapy based on the clinical situation, patient characteristics, and comorbidities, and, on the other, a combination of distinct complementary strategies. Some of these studies are already being designed and are awaiting funding.
CONCLUSIONSProtection against myocardial ischemia-reperfusion injury is a promising strategy for ameliorating the consequences of coronary disease for individual and societal health. During the coming years, it will be necessary to focus on studying the molecular mechanisms of cell death during myocardial reperfusion and on developing new therapies to prevent cell death and to establish the best way to use these treatments in clinical practice. Given that these treatments would be used just once in most patients, their development is at a disadvantage compared with drugs that require more long-term use, and which are therefore much more profitable. To date, drugs used for this purpose have been used for other indications (repurposing). The task of adding to the arsenal of specifically designed drugs combating reperfusion injury must be actively encouraged. The role of public funding is particularly crucial for researching treatments without economic value, such as RIC. The available data raise the hope that, with the combined efforts of researchers, academics, public bodies, and industry, treatment of reperfusion injury will become a reality that improves survival and quality of life in patients with STEMI and limits the social impact of ischemic heart disease in the near future.
FUNDINGThis work has been funded by the Ministry of Economy and Competition and the Instituto de Salud Carlos III (Cardiovascular Research Network RD12/0042/0021, PI12/00788, PI12/01738, and SAF2008/03067).
CONFLICTS OF INTERESTSNone declared.
Section sponsored by AstraZeneca.