Pese a recibir lo más tempranamente posible la terapia de reperfusión, un amplio número de los pacientes que sufren infarto agudo de miocardio con elevación del segmento ST tienen infartos que comprometen su supervivencia y su calidad de vida. Parte de la muerte celular secundaria a una oclusión coronaria transitoria ocurre durante la reperfusión, por mal manejo del calcio en el sistema retículo sarcoplasmático-mitocondria, activación de calpaínas, estrés oxidativo y fallo mitocondrial, favorecidos por la rápida normalización del pH intracelular. Varios ensayos clínicos han demostrado que se puede limitar el tamaño del infarto mediante estrategias no farmacológicas —como el poscondicionamiento isquémico y el condicionamiento isquémico remoto— o farmacológicas —como la estimulación de la síntesis de guanosina monofosfato cíclico, la insulina, los agonistas del péptido glucagonoide tipo 1, los bloqueadores beta o la ciclosporina. Diversos ensayos clínicos han dado resultados negativos, en la mayoría de los casos por falta de datos preclínicos consistentes o un diseño equivocado, en particular, administración tardía. Son necesarios, pues, ensayos clínicos grandes con variables clínicas primarias y terapias combinadas y que consideren edad, sexo y comorbilidades, para que la protección contra el daño por reperfusión se convierta en un tratamiento estándar para los pacientes con infarto agudo de miocardio con elevación del segmento ST.
Palabras clave
La cardiopatía isquémica es la primera causa de muerte en el conjunto de la población mundial y, si no se producen cambios de tendencia imprevistos, seguirá siéndolo en 20301. El impacto social de la cardiopatía isquémica es enorme no solo por la mortalidad que causa, sino por la morbilidad, la pérdida de calidad de vida y el elevado coste económico de todo ello. Este impacto se debe en su mayor parte a un mecanismo fisiopatológico: la muerte de los cardiomiocitos. En la cardiopatía isquémica, dicha muerte se produce casi exclusivamente en el contexto de episodios de isquemia miocárdica grave y prolongada, que ocurren como consecuencia de la complicación trombótica de placas de ateroma de las arterias coronarias epicárdicas, en lo que se conoce como síndrome coronario agudo. La muerte de los cardiomiocitos es más importante cuando la isquemia ocurre por oclusión coronaria completa, en ausencia de circulación colateral bien desarrollada, afecta a la mayor parte del espesor de la pared del ventrículo izquierdo y cursa con elevación del segmento ST en el electrocardiograma (infarto agudo de miocardio con elevación del segmento ST [IAMCEST]).
La muerte celular que ocurre en el síndrome coronario agudo no solo produce una pérdida directa de la actividad contráctil, sino que, además, puede causar cambios geométricos en la pared infartada, y cambios adaptativos en el miocardio restante, que conduzcan finalmente a disfunción general y dilatación del ventrículo, en un proceso denominado remodelado adverso2,3. La cicatriz y el remodelado adverso conducen a insuficiencia cardiaca y favorecen la aparición de arritmias ventriculares potencialmente letales4, de modo que la muerte celular ocurrida durante el síndrome coronario agudo acaba por determinar no solo la mortalidad en la fase aguda, sino la morbimortalidad a largo plazo5.
Disminuir la muerte celular durante el síndrome coronario agudo, y en particular durante el IAMCEST, parece pues una estrategia lógica para disminuir el impacto de la cardiopatía isquémica en la salud y la sociedad.
La prevención del daño por reperfusión en el conjunto de estrategias para disminuir el impacto de la cardiopatía isquémicaLa cardiopatía isquémica es una enfermedad de etiología mixta, genética y ambiental, cuyo sustrato anatomopatológico es la placa de ateroma. La ateromatosis coronaria se desarrolla de modo clínicamente silente durante años y solo produce manifestaciones clínicas cuando la luz del vaso se estrecha mucho, ya sea como consecuencia del crecimiento de la placa de ateroma o por la aparición de trombosis intracoronaria debida a la complicación de la placa, lo que da lugar a una pérdida de la continuidad endotelial por erosión, fisura o rotura del endotelio6. Por lo tanto, prevenir la aparición de placas de ateroma coronarias o su progresión es la primera línea de actuación contra la enfermedad.
Sin embargo, la prevención de la cardiopatía isquémica no está exenta de dificultades. Las intervenciones en el conjunto de la población son de gran coste y no pueden ser muy agresivas, ya que, de lo contrario, el precio en efectos adversos sería muy alto. Las intervenciones más agresivas, encaminadas a evitar el crecimiento de la placa, disminuir el riesgo de complicaciones o atenuar la trombosis secundaria, deben limitarse al tratamiento de sujetos con alto riesgo de sufrir la enfermedad. Aunque se ha avanzado mucho en la identificación de los factores de riesgo y el desarrollo de métodos para calcular el riesgo individual7, la eficacia en el control de los factores de riesgo de la enfermedad convencionales es escasa8. Por otra parte, los estudios genéticos han identificado múltiples loci asociados a la aparición de la enfermedad ateromatosa, pero esta asociación es muy débil y su efecto en la capacidad predictiva de los factores de riesgo convencionales es escasa, especialmente cuando se tiene en cuenta los antecedentes familiares. La dificultad en prevenir la cardiopatía isquémica queda clara cuando se observa la evolución de su incidencia en distintas poblaciones. En España concretamente, se está lejos de controlar la cardiopatía isquémica con medidas de prevención, teniendo en cuenta, además, que la incidencia de los factores de riesgo convencionales se mantiene esencialmente estable y en algunos casos se prevé que aumente, como ocurre con la diabetes mellitus tipo 2, favorecida por la creciente frecuencia de obesidad infantil9,10.
Es importante tener en cuenta que la eficacia de las estrategias de estratificación del riesgo y la prevención debe medirse no solo por su capacidad para disminuir la aparición de manifestaciones clínicas de cardiopatía isquémica, sino más específicamente, por su capacidad para disminuir la incidencia de síndrome coronario agudo y, en concreto, infarto agudo de miocardio (IAM), que originan la mayor parte de la morbilidad y la mortalidad asociadas a la enfermedad. Sin embargo, existe amplia evidencia de que la ocurrencia de IAM es muy difícil de predecir. En un gran estudio reciente, realizado en 542.008 pacientes con un primer infarto de miocardio, aproximadamente la mitad tenía uno o ningún factor de riesgo convencional11,12. Más aún, cuando se analizó la mortalidad hospitalaria en distintos subgrupos de pacientes, esta fue inversamente proporcional al número de factores de riesgo clásicos que presentaban11. Considerando ambas observaciones conjuntamente, se puede calcular que la gran mayoría de los pacientes que mueren en el hospital por un primer IAM presentaban un perfil de riesgo bajo, con 0–1 factores de riesgo. Por lo tanto, aunque es obvio que los esfuerzos para mejorar la estratificación del riesgo y la prevención de la cardiopatía isquémica son esenciales, no lo es menos la necesidad de mejorar la eficacia del tratamiento del IAM, de modo que su pronóstico mejore. En muchos casos, esta es la primera y única opción que tenemos de alterar el curso de la enfermedad.
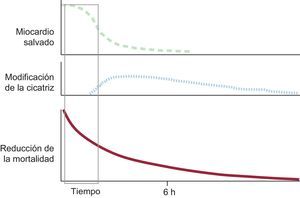
El pronóstico del IAM depende fundamentalmente de su extensión, es decir, de la cantidad de células que mueren durante el evento13. El tamaño final de una necrosis depende fundamentalmente de la velocidad de progresión del daño isquémico (influida por el flujo residual, ya sea a través de la lesión o la circulación colateral, y por la temperatura, entre otros factores) y la duración de la isquemia14,15. El tratamiento más efectivo para limitar el tamaño del infarto es la reperfusión precoz16–18. Sin embargo, la cantidad de miocardio salvado por la reperfusión disminuye rápidamente a medida que esta se retrasa y la ventana de tiempo durante la cual la reperfusión limita efectivamente el tamaño del infarto en pacientes con IAMCEST es breve. Después de 3 h de isquemia, en ausencia de circulación colateral y flujo residual (TIMI [Thrombolysis in Myocardial Infarction] 0),la cantidad de miocardio salvado es, en la mayoría de los casos, pequeña o nula19. La reperfusión más tardía también es beneficiosa, y se recomienda realizarla, en general, durante las primeras 12 h tras la aparición de los síntomas20, pero en este caso el beneficio se debe a los efectos positivos de la reperfusión para la cicatrización, que limita su expansión y el remodelado adverso secundario20 (figura 1).

El beneficio de la reperfusión se debe fundamentalmente a la limitación del tamaño del infarto cuando el tiempo total de isquemia es breve. Cuando la reperfusión es más tardía, el efecto en la cicatrización con menor expansión de la cicatriz y remodelado adverso puede ser más importante.
La breve ventana disponible para limitar el tamaño del infarto mediante la reperfusión hace que, incluso cuando se dispone de sistemas adecuados de administración rápida de tratamiento de reperfusión (angioplastia primaria o trombolisis sin pasar por urgencias, antiagregación y anticoagulación óptimas, etc.), este no evite, en la gran mayoría de los pacientes, la aparición de necrosis que afecta a la mayor parte del espesor de la pared ventricular, lo que produce ondas Q, con consecuencias desfavorables para el pronóstico. La visión convencional es que, cuando el paciente con IAMCEST llega a la sala de cateterismo, como ya no es posible acortar la duración de la isquemia ni modificar la circulación colateral o el flujo coronario residual (flujo TIMI), lo único que se puede hacer es abrir la arteria de manera completa y definitiva y observar si se ha llegado a tiempo de salvar miocardio. Sin embargo, una enorme cantidad de información preclínica y una creciente cantidad de datos clínicos indican que es posible aumentar la proporción de miocardio salvado por la reperfusión mediante tratamientos aplicados más o menos en el momento de la reperfusión. El presente artículo analiza la situación actual y el futuro esperable de los tratamientos encaminados a disminuir la muerte celular de pacientes con IAMCEST que reciben tratamiento de reperfusión, y no analizará, por lo tanto, la eficacia de estos tratamientos en otros contextos clínicos, como la cirugía cardiaca o la reanimación cardiopulmonar.
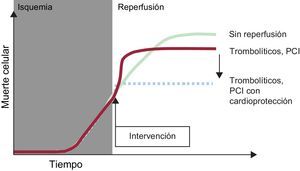
DAÑO POR REPERFUSIÓNConceptoLos primeros experimentos de laboratorio con oclusión coronaria transitoria permitieron observar que durante la reperfusión se producía una paradójica exacerbación de las alteraciones funcionales asociadas a la isquemia, en particular arritmias21–23. Estudios en corazones aislados y perfundidos demostraron un aumento en la liberación de enzimas cardiacas al restaurar el flujo después de un periodo de isquemia24. Posteriormente, se comprobó que, incluso en ausencia de necrosis, la reperfusión se acompañaba de disfunción contráctil transitoria (aturdimiento)25. Sin embargo, las diferencias en los mecanismos y la relevancia clínica de todos estos fenómenos asociados a la reperfusión dificultaron la aceptación inicial del concepto de daño por reperfusión entre la comunidad científica y médica. La situación cambió cuando se introdujo el concepto de daño letal por reperfusión, con una definición operativa precisa: la muerte celular que se puede prevenir mediante intervenciones aplicadas en el momento de la reperfusión26. En este artículo utilizaremos siempre la expresión «daño por reperfusión» según esta definición, cuya aplicación en el caso del IAMCEST se ilustra en la figura 227.

Ilustración del concepto de daño por reperfusión. Durante la isquemia miocárdica, la muerte celular progresa. La reperfusión detiene este proceso, pero el beneficio se limita por el aumento de la muerte celular durante los primeros minutos de reperfusión; esto se puede evitar con tratamientos cardioprotectores aplicados en el momento de la restauración del flujo. PCI: intervención coronaria percutánea primaria. Modificada con permiso de Garcia-Dorado et al27.
La oclusión de una arteria coronaria crea una zona de isquemia (área en riesgo) cuyos bordes laterales están nítidamente definidos debido a la ausencia de comunicación entre lechos capilares adyacentes28,29 y en la que el flujo residual, si lo hay, se distribuye por el subepicardio. La reperfusión se acompaña de edema extracelular e intracelular, debido a la permeabilización endotelial y la creación de un gradiente osmótico entre los espacios extravascular e intravascular30. El edema extracelular aparece a los pocos minutos de la reperfusión y puede contribuir a las alteraciones de la función mecánica del miocardio y durar varios días en pacientes con IAMCEST, de modo que se ha utilizado para delimitar la zona de área en riesgo mediante resonancia magnética30. Además de edema, la reperfusión se acompaña de depósito de plaquetas en todo el miocardio reperfundido como consecuencia de su adhesión (dependiente de P-selectina) al endotelio activado.
Si la reperfusión no se lleva a cabo de forma suficientemente precoz, se produce una zona de muerte celular. La muerte celular ocurre fundamentalmente por necrosis, y muy rápidamente, en los primeros minutos de reperfusión26. En contra de lo que se pensó durante un tiempo, la apoptosis no tiene un papel importante en la muerte por isquemia-reperfusión; de hecho, los cardiomiocitos adultos tienen silenciada la expresión de proteínas esenciales en el programa de apoptosis31. La zona necrótica tiene una extensión lateral que coincide con la extensión lateral del área en riesgo, y una extensión transmural que crece hacia el subepicardio a medida que la reperfusión se retrasa (fenómeno del frente de onda)14. Cuando la reperfusión es tardía y el infarto es extenso, se producen en su interior zonas de daño microvascular grave con pérdida de la barrera endotelial, hemorragia intersticial y detención del flujo (áreas sin reflujo)32. Las áreas sin reflujo se asocian a infartos extensos y mal pronóstico, lo que ha llevado a pensar que pueden contribuir a la muerte celular. Sin embargo, no hay evidencia definitiva de que esto sea así, y muchos datos indican que las zonas sin reflujo se producen en zonas ya necrosadas33. Lo que sí parece probable es que las zonas sin reperfusión dificulten la cicatrización y favorezcan la expansión de la cicatriz y el remodelado adverso posterior34.
Daño celular isquémicoEl daño por reperfusión, así definido, puede ocurrir en diversas situaciones clínicas, aparte del IAMCEST, en las que se produce muerte celular secundaria a isquemia miocárdica transitoria, en particular durante la cirugía cardiaca o en la reanimación cardiopulmonar. Sin embargo, los mecanismos implicados pueden ser diferentes en función de las alteraciones producidas antes de restaurar el flujo (concomitancia de hipotermia, soluciones cardiopléjicas, hipoxia con flujo preservado, etc.). Nos centraremos en los mecanismos del daño por reperfusión que aparece después de un periodo de isquemia grave regional y normotérmica.
Se ha revisado en múltiples ocasiones las alteraciones celulares producidas por la isquemia26,35–38. La isquemia regional causa, en pocos segundos, la detención de la fosforilación oxidativa debido a la falta de oxígeno en la cadena respiratoria mitocondrial. La síntesis de adenosintrifosfato (ATP) queda limitada a la vía glucolítica, lo cual conduce, con el ciclo de Krebs detenido, a una acumulación intracelular de ácido láctico26,38. Este hecho, junto con la imposibilidad de eliminar el CO2 del espacio extracelular por la detención del flujo, hace que el pH intracelular caiga rápidamente hasta alcanzar en pocos minutos valores próximos a 6,439,40. La acidosis y la acumulación de fosfato inorgánico detienen en pocos segundos la actividad contráctil, incluso cuando la concentración de ATP sigue siendo prácticamente normal. Durante los minutos siguientes, la concentración de ATP sigue descendiendo hasta alcanzar niveles críticos, muy próximos a cero, que promueven la aparición del rigor isquémico41. El agotamiento del ATP se acelera en su fase final por la inversión de la actividad de la ATPasa mitocondrial (complejo V de la cadena respiratoria), que lo consume para mantener el potencial de membrana mitocondrial42. La acidosis activa el intercambiador Na+/H+ del sarcolema, con la consiguiente entrada de Na+, mientras que el agotamiento energético detiene la expulsión de Na+ a través de la Na+/K+ ATPasa. Como consecuencia de todo ello, el Na+ se acumula en el interior de la célula y hace funcionar el intercambiador Na+/Ca2+ en sentido inverso y aumenta el Ca2+ citosólico26,38. La célula se despolariza. Las mitocondrias captan Ca2+ a favor de la electronegatividad de su matriz al inicio de la isquemia, pero dejan de hacerlo cuando se despolarizan43. El retículo sarcoplasmático tampoco puede captar Ca2+ del citosol, ya que el transportador necesita ATP para funcionar.
Daño por reperfusiónLa reinstauración del flujo miocárdico hace que se recupere la actividad respiratoria, el potencial de membrana mitocondrial y la síntesis de ATP. El potencial de membrana celular se recupera, lo que, en presencia de una concentración elevada de Na+ en el citosol, pone en marcha el intercambiador Na+/Ca2+ en su forma inversa, lo que agrava aún más la sobrecarga de Ca2+ citosólico. La disponibilidad de ATP en presencia de Ca2+ aumentado activa la captación de Ca2+ por el retículo sarcoplasmático hasta superar el umbral necesario para abrir los canales de rianodina, con lo que el Ca2+ se libera al citosol. La repetición cíclica de este proceso da lugar a oscilaciones de Ca2+ que se propagan por la célula26,38. Los canales de rianodina están conectados físicamente con las mitocondrias, de modo que el Ca2+ se libera específicamente en microdominios subcelulares desde los que puede entrar, a favor del gradiente de potencial, a la matriz mitocondrial a través del unitransportador mitocondrial de Ca2+. Las características fisicoquímicas de este transportador hacen que las oscilaciones de Ca2+ favorezcan el aumento del Ca2+ mitocondrial. Las oscilaciones de Ca2+ y su efecto en las mitocondrias dependen, por una parte, de la actividad de los canales de rianodina y el unitransportador mitocondrial de Ca2+ y, por otra, de la actividad del transportador encargado, que a su vez se modula por fosfolambano44–46. Durante la reperfusión, la activación de la proteincinasa A, la proteincinasa G (PKG) y la oxidación/fosforilación de la cinasa dependiente de calcio y calmodulina modulan, a través de cambios en la fosforilación de los canales de rianodina, fosfolambano y el unitransportador mitocondrial de Ca2+, la magnitud de las oscilaciones de Ca2+ y el grado de sobrecarga de Ca2+ mitocondrial47.
Las oscilaciones citosólicas de Ca2+ tienen consecuencias deletéreas para las células. En primer lugar, favorecen una activación contráctil excesiva, que puede desencadenar hipercontractura48. La hipercontractura es capaz romper la arquitectura celular y causar rotura sarcolemal y muerte celular49. En segundo lugar, las oscilaciones de Ca2+ crean, a su vez, oscilaciones del potencial de membrana celular en forma de despolarizaciones precoces o pospotenciales, un mecanismo importante de las arritmias por reperfusión50. En tercer lugar, favorecen la apertura del poro de transición mitocondrial (mPTP).
El mPTP es un canal de alta permeabilidad, de identidad molecular no bien determinada, que permite la conexión directa entre la matriz mitocondrial y el espacio intermembranario51. Al abrirse, disipa el potencial de membrana mitocondrial y detiene la síntesis de ATP, causa edema y rotura mitocondrial y permite la salida de moléculas mitocondriales al citosol, incluido el Ca2+. Se considera que la permeabilización es un mecanismo importante de muerte celular durante la reperfusión. La permeabilización mitocondrial y la hipercontractura secundaria a la sobrecarga y las oscilaciones de Ca2+ están interrelacionadas, de modo que las oscilaciones favorecen la apertura del mPTP52 y la permeabilización mitocondrial promueve la salida de Ca2+ hacia el citosol, lo que favorece el desarrollo de más oscilaciones e hipercontractura53. Parece que la hipercontractura tiene un papel más importante en la génesis de la muerte celular después de episodios de isquemia relativamente breves, mientras que el mPTP es el principal mecanismo de la muerte celular por reperfusión después de una isquemia prolongada54.
Tanto la hipercontractura como la apertura del mPTP se ven favorecidas por dos fenómenos importantes que ocurren durante los primeros minutos de reperfusión. El primero es la normalización rápida del pH intracelular, que permite la activación contráctil y la apertura del mPTP, ambas inhibidas por la acidosis55,56. Además, la normalización del pH permite la activación de las calpaínas, proteasas dependientes de Ca2+ que se encontraban inhibidas por el pH ácido. La activación de las calpaínas daña el citoesqueleto e impide la función normal de la Na+/K+ ATPasa, con lo que agrava la sobrecarga de Na+ y Ca2+ y cierra el círculo vicioso57–59.
Durante la reperfusión se generan radicales libres del oxígeno que participan de manera muy importante en el daño por reperfusión, ya que favorecen directamente la apertura del mPTP60 y agravan las alteraciones de la homeostasis del Ca2+ oxidando la cinasa dependiente de calcio y calmodulina61. Además, lo que es importante, el anión superóxido disocia la sintasa del óxido nítrico (NO) de la tetrahidrobiopterina, lo que resulta en la producción de anión superóxido, un potente oxidante, en vez de NO. El resultado es la disminución de la disponibilidad de NO y de la vía de señalización que este inicia (guanosina monofosfato cíclico [GMPc] y PKG)62. La disminución de PKG acelera la normalización del pH (ya que la PKG inhibe el intercambiador Na+/H+) y favorece las oscilaciones del Ca2+ (actuando sobre fosfolambano), acciones ambas con efectos negativos para la supervivencia celular, como ya se ha explicado.
Finalmente, la muerte celular puede propagarse entre miocitos adyacentes a través de las comunicaciones intercelulares tipo gap junctions63–66, que permiten el paso de Na+ de una célula a otra67. El Na+, a su vez, favorece la entrada de Ca2+ en la célula contigua y puede causarle la muerte si está suficientemente dañada (es decir, si la función de la bomba de Na está alterada, ha daño en el citoesqueleto, etc.)67. La comunicación intercelular a través de gap junctions también requiere la normalización del pH, puesto que la acidosis provoca el cierre de estos canales68.
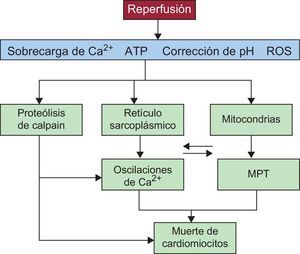
Los mecanismos mencionados se resumen en la figura 3.
 (véase la explicación detallada en el texto). ATP: adenosintrifosfato; MPT: transición de permeabilidad mitocondrial; ROS: radicales de oxígeno libres.")
Esquema de los principales mecanismos de la muerte de los cardiomiocitos durante la reperfusión miocárdica. Durante la reperfusión se producen oscilaciones de calcio dependientes del retículo sarcoplasmático y permeabilización mitocondrial favorecida por la sobrecarga de calcio y el estrés oxidativo. La recuperación de la acidosis favorece ambos fenómenos, íntimamente relacionados gracias al contacto físico entre mitocondrias y retículo sarcoplasmático y también la activación de proteasas dependientes del calcio (calpaína) (véase la explicación detallada en el texto). ATP: adenosintrifosfato; MPT: transición de permeabilidad mitocondrial; ROS: radicales de oxígeno libres.
Tanto la isquemia como la reperfusión miocárdica suceden, en realidad, en un tejido que contiene un número muy alto de células que no son miocitos, como células endoteliales, fibroblastos y células sanguíneas. Las plaquetas activadas que se depositan en el miocardio reperfundido tienen un efecto deletéreo para los cardiomiocitos, por mecanismos independientes de la obstrucción del flujo microvascular y probablemente relacionados con la liberación de sustancias como la trombina. De este modo son capaces de empeorar el manejo del Ca2+ y desencadenar otros efectos adversos en las células69–72. La activación de las células endoteliales y la liberación de micropartículas y ARN ribosómico también afecta negativamente a la supervivencia de los miocitos a través de la activación masiva de factor de necrosis tumoral alfa, entre otros factores73.
ESTRATEGIAS MÁS PROMETEDORAS PARA LIMITAR EL DAÑO POR REPERFUSIÓNLa demostración de la existencia del daño por reperfusión se basó en la utilización de herramientas farmacológicas que interferían con los mecanismos antes descritos. Sin embargo, por razones que se analizan más adelante, ninguna de estas herramientas se ha trasladado a la práctica clínica. Por lo tanto, son múltiples las dianas potencialmente útiles para el tratamiento del daño por reperfusión para las que no se han desarrollado medicamentos74.
Cardioprotección endógenaEl primer tratamiento aplicado con éxito a pacientes con IAMCEST fue el poscondicionamiento isquémico (PostC)75,76. A diferencia de los tratamientos farmacológicos, el PostC disminuye el daño por reperfusión introduciendo breves episodios de isquemia justo al comienzo de la reperfusión77, por lo cual tiene efecto protector sin necesidad de agentes externos (cardioprotección endógena).
El PostC actúa por mecanismos diferentes que el precondicionamiento isquémico, en el que periodos de isquemia breves y repetidos inmediatamente antes de un episodio largo disminuyen la muerte celular causada por este. El precondicionamiento isquémico ofrece una protección más robusta, pero no es aplicable a pacientes con IAMCEST. La protección otorgada por el PostC no solamente es menor, sino que varía dependiendo de las condiciones. Su eficacia es escasa o nula después de episodios de isquemia breves que causan infartos pequeños78. El efecto protector del PostC se debe fundamentalmente a que retrasa unos minutos la normalización del pH intracelular, debido al enlentecimiento del lavado de metabolitos secundario a las interrupciones del flujo, y a la disminución del daño oxidativo, que preserva la vía de señalización NO-GMPc-PKG, inhibiendo el el intercambiador Na+/H+79,80.
Recientemente se ha descrito una forma de protección endógena consistente en inducir a distancia (generalmente en las extremidades) isquemia miocárdica intermitente inmediatamente antes o en los primeros minutos de reperfusión: el condicionamiento isquémico remoto (RIC)81–83. El RIC tiene sobre el PostC la ventaja de que no requiere manipular la arteria coronaria durante los primeros minutos de reperfusión, pero los mecanismos por los que ejerce su efecto protector siguen sin esclarecerse83,84.
Cardioprotección farmacológicaLa preservación de la vía de señalización de la PKG tiene múltiples efectos protectores. Además de retrasar la normalización del pH y reducir las oscilaciones del Ca2+ como ya se ha descrito, parece que tiene efectos mitocondriales directos que dificultan la permeabilización mitocondrial85. De hecho, hay amplia evidencia del efecto protector de los tratamientos que normalizan el GMPc al principio de la reperfusión, bien sea mediante estimulación de la guanilatociclasa particulada mediante péptidos natriuréticos86, bien mediante estimulación directa e independiente de NO de la guanilatociclasa soluble87. La utilidad de la estimulación de la guanilatociclasa soluble mediante NO exógeno no es tan homogénea, probablemente porque el exceso de NO puede causar daño oxidativo88.
Otra estrategia ampliamente estudiada, usando tanto intervenciones farmacológicas89,90 como modelos genéticamente modificados90, es inhibir la apertura del mPTP. En ambos casos se actúa sobre una de las proteínas que participan en su apertura inducida por Ca2+, la ciclofilina D. La ciclosporina, un fármaco ampliamente utilizado como inmunosupresor, actúa a través de la ciclofilina D inhibiendo el mPTP89,90 en respuesta al Ca2+. Se está estudiando otras moléculas para inhibir el mPTP sobre las que la información existente es preliminar. Más recientemente, se ha identificado la disfunción del complejo I de la cadena respiratoria como una diana potencialmente útil, y se han propuesto distintas formas de prevenirla, sobre las que, sin embargo, hay poca información.
Se están realizando estudios preclínicos sobre otras dianas, entre las que destacan las relacionadas con la vía de señalización de la insulina y el metabolismo energético. Entre las primeras, la aplicación precoz de soluciones con glucosa, insulina y potasio91–93 y los miméticos estables del péptido glucagonoide tipo 194 han recibido particular atención y han proporcionado resultados prometedores. Otras formas potencialmente protectoras de alterar el metabolismo energético en el momento de la reperfusión incluyen el uso de trimetazidina95 o el aporte de sustratos específicos96.
Finalmente, hay muchas otras intervenciones sobre distintas dianas dirigidas a mecanismos del daño por reperfusión; sin embargo, sus efectos resultan controvertidos. Entre ellas se encuentra la activación de leucocitos o factores del complemento (que ocurre cuando la mayor parte de la muerte celular ha sucedido) o la apoptosis dependiente de caspasas (una vía de muerte celular prácticamente inoperante en cardiomiocitos adultos).
ENSAYOS CLÍNICOSDurante la última década, varios ensayos clínicos han testado en pacientes distintas estrategias para limitar el tamaño del infarto y disminuir el daño por reperfusión. Sus poblaciones diana, tamaños muestrales, diseños, objetivos primarios y resultados se han revisado recientemente97–100. Todos ellos carecen de potencia estadística para detectar efectos en la evolución clínica y están dimensionados para detectarlas en variables subrogadas, en concreto tamaño del infarto, medido o estimado por distintos métodos. Algunas estrategias, como el PostC, han sido objeto de múltiples estudios e incluso metanálisis, mientras que para otras intervenciones solo hay datos disponibles de un único estudio. A continuación se analizan los resultados obtenidos con distintas intervenciones cardioprotectoras.
Condicionamiento isquémicoEn general, los estudios basados en maniobras de condicionamiento isquémico han sido positivos101.
Poscondicionamiento isquémicoVarios estudios han analizado la eficacia de este tratamiento, el primero descrito como capaz de limitar el daño por reperfusión de los pacientes. En el estudio inicial, se realizaron reoclusiones de la arteria reperfundida mediante cuatro inflados del balón de angioplastia de 1 min de duración, separados por periodos de 1 min con el balón desinflado, comenzando al minuto de abrir la arteria. Este protocolo disminuyó significativamente el tamaño del infarto medido como área bajo la curva de creatincinasa75. Estudios posteriores realizados por distintos grupos obtuvieron resultados variados76,102–105, cuyas diferencias se ha intentado explicar por la participación de distintos factores, como el diferente grado de utilización de stent directo, o diferencias en la antiagregación perioperatoria103. Aunque considerados en general los datos existentes indican que el PostC produce cierta disminución del tamaño del infarto106, el uso de este procedimiento no se ha extendido apreciablemente durante los últimos años, probablemente por la escasa y variable protección que parece proporcionar, su carácter antiintuitivo (producir reoclusiones) y el temor a complicaciones por los inflados repetidos del balón de angioplastia. Una limitación común a los distintos ensayos clínicos sobre PostC es que no pueden ser a doble ciego. Esta es una limitación importante que deja abierta la puerta a posibles sesgos en la exclusión de pacientes, asignación de eventos y determinaciones no completamente automatizadas, como es el caso de la determinación del tamaño mediante resonancia magnética107.
Condicionamiento isquémico a distanciaAl igual que ocurrió con el PostC, el RIC se aplicó a pacientes al poco tiempo de haberse descrito en estudios de laboratorio y mucho antes de que sus mecanismos fuesen conocidos108. El RIC puede inducirse mediante el inflado de un manguito de presión arterial colocado en el brazo hasta presiones superiores a la sistólica durante periodos de 5 min separados 5 min. Los resultados descritos hasta el momento son coherentemente positivos y concordantes con la evidencia preclínica. El RIC tiene múltiples ventajas sobre el PostC, y es un firme candidato a ocupar un lugar en las recomendaciones de práctica clínica. En primer lugar, es extraordinariamente seguro y barato; segundo, puede aplicarse en la ambulancia y a todos los pacientes con IAMCEST que reciben tratamiento de reperfusión, no solo a los que reciben angioplastia primaria, y tercero, la protección proporcionada por el RIC parece ir más allá del área miocárdica en riesgo y el IAMCEST. Hay en marcha estudios en otros tejidos109, y ya hay evidencia sólida de que disminuye el daño miocárdico en distintos tipos de cirugía110,111, particularmente la revascularización coronaria112,113. Aunque es complicado hacer que los estudios sobre RIC sean a doble ciego, no es imposible, ya que se puede utilizar inflados a baja presión programados automáticamente que actúan como tratamiento placebo114.
Tratamientos farmacológicosLos resultados de tratamientos farmacológicos para disminuir el tamaño del infarto en IAMCEST son tan variados como las estrategias y los diseños utilizados. Sin embargo, los estudios basados en estrategias con una experiencia preclínica claramente sólida y diseños que reproducen fielmente las condiciones de los estudios preclínicos han sido generalmente positivos108,115–119.
Péptido natriurético auricularLa diana farmacológica para la prevención del daño miocárdico por reperfusión sobre la que hay evidencia preclínica más sólida probablemente sea la vía de señalización GMPc/PKG. Sin embargo, solo un ensayo clínico ha evaluado directamente esta estrategia en patientes con IAMCEST115, y demostró limitación del infarto enzimático y mejoría de la función ventricular de pacientes que recibieron péptido natriurético auricular intravenoso en el momento de la reperfusión, de modo que reprodujeron el efecto beneficioso de este tratamiento en animales86.
Glucosa-insulina-potasioLa administración de insulina también señaliza a través de la vía GMPc/PKG (vía Akt y PI3P), además de incrementar la utilización de la glucosa120,121. La evidencia preclínica sobre la utilidad de la insulina al principio de la reperfusión miocárdica es sólida91,120-123. Sin embargo, la traslación a la clínica dio lugar a resultados negativos, fáciles de explicar por la administración demasiado tardía del tratamiento124–126. Un reciente estudio aleatorizado y a doble ciego ha encontrado, no obstante, que la administración de glucosa-insulina-potasio es capaz de reducir el tamaño del infarto y las complicaciones eléctricas graves cuando se administra en la ambulancia a pacientes con IAMCEST que van a recibir angioplastia primaria117.
Péptido glucagonoide tipo 1La estimulación de la vía del péptido glucagonoide tipo 1también actúa, entre otras vías, por la del GMPc/PKG127,128. Hay evidencia sólida sobre los efectos cardioprotectores de potenciar la señalización dependiente del péptido glucagonoide tipo 1, bien sea utilizando análogos estables del péptido glucagonoide tipo 1, bien inhibidores de su degradación128,129. Se han realizado dos ensayos clínicos para evaluar el efecto de exenatida en pacientes con IAMCEST sometidos a angioplastia primaria, y ambos han sido positivos118,130. Dado su elevado perfil de seguridad, esta estrategia resulta especialmente prometedora.
AdenosinaLa adenosina es producto de la degradación de adenosina monofosfato y tiene, entre otros, un potente efecto vasodilatador. Además de sus efectos en la vasculatura y los leucocitos, tiene efectos directos en los cardiomiocitos, pues eleva la disponibilidad de NO (vía Akt) y, por lo tanto, la vía GMPc/PKG. Se empleó por vía intravenosa en los estudios AMISTAD131, sin resultados concluyentes, aunque con indicios de protección a dosis altas en pacientes con infartos anteriores132. Después se propuso utilizarlo en forma de inyección intracoronaria previa a la reperfusión133. El estudio más grande realizado hasta la fecha es el PROMISE, un estudio a doble ciego que ha incluido a 200 pacientes con flujo TIMI 0-1 y cuya variable principal es el tamaño del infarto medido por resonancia magnética, y con un subanálisis preespecificado según la duración de la isquemia. Los resultados muestran que la adenosina no limita el tamaño del infarto en el conjunto de los pacientes, aunque sí lo hace en quienes reciben la angioplastia primaria durante las primeras 3 h desde el comienzo del dolor134, e indican que mejora la evolución de la fracción de eyección del ventrículo izquierdo durante los 6 meses siguientes al infarto (por resonancia magnética). La adenosina intracoronaria es un tratamiento barato y seguro, por lo que su uso en pacientes reperfundidos precozmente podría implementarse fácilmente si estudios posteriores, basados en variables de evolución clínica, confirman el beneficio.
CiclosporinaEn un primer estudio piloto, la ciclosporina A limitó el tamaño del infarto (determinado enzimáticamente y mediante resonancia magnética) en 58 pacientes con IAMCEST sometidos a angioplastia primaria119. Sin embargo, un estudio reciente, con un número de pacientes parecido, no encontró beneficio135. Próximamente concluirá un estudio internacional con más de 1.600 pacientes que presumiblemente establecerá la utilidad de este tratamiento para mejorar la evolución clínica de los pacientes con IAMCEST136.
Bloqueadores betaLa evidencia preclínica sobre el uso de bloqueadores beta para reducir el tamaño del infarto es mixta. Un estudio reciente encontró que la inyección intravenosa de metoprolol fue protectora en un modelo porcino sometido a 90 min de oclusión coronaria137. Este tratamiento se ha trasladado a pacientes en el estudio METOCARD-CNIC138. Se trata de un estudio con enmascaramiento que pretendía probar el efecto del metoprolol intravenoso, administrado inmediatamente antes de la angioplastia primaria, en el tamaño del infarto determinado mediante resonancia magnética; demostró protección por el aumento del miocardio salvado138,139 sin complicaciones mayores. Debe tenerse en cuenta que los bloqueadores beta ya están recomendados para los pacientes con IAMCEST. Los datos de este estudio permitirían avanzar fácilmente el momento de administrarlo, siempre que en estudios posteriores se confirmen los resultados.
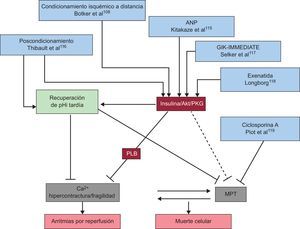
La figura 4 ilustra algunos de las más prometedoras estrategias contra el daño por reperfusión en pacientes con IAMCEST.

Algunas de las estrategias más prometedoras contra el daño por reperfusión, según los mecanismos de muerte celular con los que interfieren. ANP: péptido natriurético auricular; GIK: glucosa-insulina-potasio; MPT: transición de permeabilidad mitocondrial; pHi: pH intracelular; PKG: proteincinasa G; PLB: fosfolambano.
La aplicación de tratamientos cardioprotectores en la práctica clínica ha dado lugar, en diversas ocasiones, a estudios negativos.
En la gran mayoría de los casos, el resultado negativo era claramente predecible por los conocimientos existentes. Las razones que más frecuentemente han conducido a resultados negativos son las siguientes: a) utilización de tratamientos que, según la investigación preclínica, requieren administrarlos antes de la isquemia, como los inhibidores del intercambiador Na+/H+, que se han demostrado eficaces solo cuando se administran antes de la isquemia140–142 y fallaron en los estudios clínicos en que se administraron en el momento de la reperfusión143; b) comienzo tardío del tratamiento, pues múltiples ensayos han iniciado la aplicación de los tratamientos en estudio después de los primeros 30 min de reperfusión, cuando la mayor parte del daño por reperfusión ya ha ocurrido, como es el caso de estudios con glucosa-insulina-potasio124 o eritropoyetina144, y c) estrategias terapéuticas con evidencia preclínica claramente insuficiente o contradictoria, por ejemplo, la asistencia mecánica ventricular durante la reperfusión145, la inhibición de la proteincinasa C-delta146, la inhibición del sistema del complemento147 o el nicorandil115.
Además de un diseño incorrecto, poco justificado o muy arriesgado, otros factores pueden dificultar la traslación de los datos experimentales a la clínica100. Uno muy importante es la edad avanzada y las comorbilidades de los pacientes, en contraste con la utilización de animales sanos y jóvenes en los estudios de laboratorio. Otro es la concomitancia de terapias que pueden tener efecto protector o interferir con el de los tratamientos97. Finalmente, la elevada variabilidad del tamaño del infarto y el flujo residual o la circulación colateral en pacientes con IAM hace más difícil la detección de diferencias entre grupos97,100. Para evitar estos problemas, se han propuesto directrices respecto a la investigación preclínica y el diseño de estudios clínicos sobre protección contra el daño por reperfusión19.
Finalmente, la evidencia de que una intervención disminuye el tamaño del infarto en pacientes con IAMCEST no es suficiente para su aplicación en la práctica clínica. Es preciso realizar estudios con variables principales clínicas robustas, que requieren un gran número de pacientes con beneficio clínico confirmado.
PROTECCIÓN CONTRA EL DAÑO POR REPERFUSIÓN EN LA PRÁCTICA CLÍNICA ACTUAL Y EN EL FUTURO INMEDIATOTodos los tratamientos contra el daño por reperfusión en el IAMCEST descritos hasta aquí son en este momento experimentales. Sin embargo, se puede hablar de protección contra el daño miocárdico en algunas situaciones de la práctica clínica. En primer lugar, en estudios preclínicos se ha demostrado que algunos de los tratamientos que reciben los pacientes con IAMCEST tienen un efecto cardioprotector. Entre ellos, la morfina y sus derivados148, inhibidores P2Y12 otros fármacos antiplaquetarios149 y las estatinas150 o los bloqueadores beta137. En segundo lugar, hay tratamientos que, por su simplicidad y seguridad, algunos grupos podrían adoptar antes de que haya evidencia sólida sobre su utilidad clínica real. El PostC es uno de ellos. El RIC podría entrar en esta categoría.
En este momento hay estudios en marcha para evaluar tratamientos contra el daño por reperfusión utilizando variables subrogadas, y están ya en marcha algunos grandes estudios que utilizan variables de resultado clínico. La siguiente frontera será probablemente doble; por una parte, incluirá la personalización del tratamiento antirreperfusión en función de la situación clínica, las características del paciente y las comorbilidades. Por otra, la combinación de distintas estrategias complementarias. Algunos de estos estudios ya están en fase de diseño y pendientes de financiación.
CONCLUSIONESLa protección miocárdica contra el daño por reperfusión es una estrategia prometedora para disminuir las consecuencias de la enfermedad coronaria para la salud individual y la sociedad. Durante los próximos años será necesario profundizar en el estudio de los mecanismos moleculares de la muerte celular durante la reperfusión miocárdica y en el desarrollo de nuevos tratamientos para prevenirlo y definir la mejor forma de utilizar estos tratamientos en la práctica clínica. Al tratarse de tratamientos que se aplicarían una sola vez en la mayoría de los pacientes, su desarrollo compite en desventaja con el de fármacos para uso crónico, mucho más rentables. Hasta ahora, los fármacos utilizados para este fin ya se estaban utilizando en otras indicaciones (repurposing). La tarea de añadir al arsenal contra el daño por reperfusión fármacos específicamente diseñados se debería incentivar activamente. El papel de la financiación pública es particularmente imprescindible en la investigación de tratamientos sin valor económico, como el RIC. Los datos disponibles hacen esperar que, con el esfuerzo combinado de investigadores, académicos, agencias públicas e industria, el tratamiento del daño por reperfusión sea una realidad que mejore la supervivencia y la calidad de vida de los pacientes con IAMCEST y limite el impacto social de la cardiopatía isquémica en un futuro próximo.
FINANCIACIÓNEste trabajo ha sido financiado por el Ministerio de Economía y Competitividad y el Instituto de Salud Carlos III (Red de Investigación Cardiovascular RD12/0042/0021, PI12/00788, PI12/01738 y SAF2008/03067).
CONFLICTO DE INTERESESNinguno.
Sección patrocinada por AstraZeneca.