In the last decade, proteomics and metabolomics have contributed substantially to our understanding of cardiovascular diseases. The unbiased assessment of pathophysiological processes without a priori assumptions complements other molecular biology techniques that are currently used in a reductionist approach. In this review, we highlight some of the “omics” methods used to assess protein and metabolite changes in cardiovascular disease. A discrete biological function is very rarely attributed to a single molecule; more often it is the combined input of many proteins. In contrast to the reductionist approach, in which molecules are studied individually, “omics” platforms allow the study of more complex interactions in biological systems. Combining proteomics and metabolomics to quantify changes in metabolites and their corresponding enzymes will advance our understanding of pathophysiological mechanisms and aid the identification of novel biomarkers for cardiovascular disease.
Keywords
Cardiovascular disease (CVD) is the leading cause of mortality and morbidity in industrialized countries. Prediction of cardiovascular events relies on monitoring conventional risk factors such as age, sex, smoking habits, diabetes, and hypertension.1 Many of these risk factors are highly prevalent in the population, and even the best algorithms for acute coronary events fail to predict the majority of cases of CVD over a 10-year period.2 In addition to the difficulties of prediction, CVDs such as heart failure may represent a heterogeneous spectrum of etiologies, pathological stages, and genetic backgrounds. New biomarkers are urgently needed to stratify patients and personalize treatments. Currently, biomarker assessment is based on the quantification of a few proteins or metabolites.3 High throughput platforms such as proteomics and metabolomics can offer simultaneous readouts of hundreds of proteins and metabolites. In this review, we summarize the proteomics and metabolomics platforms that are currently applied in cardiovascular research and that may lead to the identification of new biomarkers with clinical utility.
PROTEOMICSThe first complete sequence of the human genome was published in 2001. Unexpectedly, this sequence contained only about 20 000 to 25 000 open reading frames that encode proteins.4 However, gene products are subject to alternative splicing and RNA editing, resulting in a variety of different protein isoforms.5 The objective of proteomics is to interrogate the proteome.6 The proteome encompasses the entire set of proteins expressed by a cell, tissue or organism, including their posttranslational modifications. Proteomics is the global analysis of gene expression using a variety of techniques to identify and characterize proteins. The term proteomics was first coined by Marc Wilkins in 1994 in analogy to genomics, the analysis of genes.
The first proteomic techniques were developed in the 1970s7 and their use has continuously evolved. It was not until the mid-1990s that proteomics was applied to the study of CVDs, with the pioneer studies of Knecht et al.8 and Jungblut et al.9 At that time, a major hurdle was the identification of proteins. Initially, Edman sequencing was used but this technique has been superseded by the advent of biological mass spectrometry (MS). The first Nobel Prize for MS was awarded to F.W. Aston in 1920. His mass spectrometer allowed separation of different isotopes. More recently, two inventions made it possible to analyse biomolecules (DNA, peptides, proteins) by MS: in 1987, M. Karas and F. Hillenkamp invented MALDI (matrix-assisted laser desorption/ionization). In MALDI-MS, a matrix (eg, α-cyano-4-hydroxycinnamic acid) is mixed with an analyte (eg, peptides). The analyte is desorbed from the matrix with a laser shot and is ionized.10 In 1989, J.B. Fenn invented electrospray ionization.11 He was awarded the Nobel Prize in Chemistry in 2002. In electrospray ionization, the analyte is ionized from a liquid phase into the gas phase. Thus, liquid chromatography (LC) systems could be directly interfaced to mass spectrometers. LC-tandem MS (LC-MS/MS) is the current gold standard in proteomics. LC first separates peptides, which is essential because most mixtures are far too complex to be analyzed by MS without prefractionation. The tandem mass spectrometer then records the masses of the intact peptides (full MS) before one precursor ion is selected and fragmented. Fragmentation is commonly induced by collision with argon or nitrogen. More recent methods also use electrons, a softer fragmentation method that preserves posttranslational modifications.12 The fragments are recorded in an MS/MS spectrum and the fragmentation pattern reveals a specific Δ mass for each amino acid in the peptide. Most early proteomics workflows were based on an initial step of protein separation, which involved techniques such as gel electrophoresis. As the mass spectrometers became faster and more sensitive, protein mixtures of increasing complexity were digested and analyzed directly without prior fractionation at the protein level. The latter method is termed bottom-up proteomics. In contrast, top-down proteomics analyses intact proteins by MS. However, this method is still limited to single proteins in solutions.
Tandem Mass SpectrometryThe bottom-up techniques are currently the workhorse to analyze biological samples: in “shotgun proteomics”, proteins in complex mixtures are analyzed using a combination of high-performance LC and MS/MS. A potential caveat of this technique is that the mass spectrometer selects the most abundant precursor ion for fragmentation. Thus, abundant proteins are more likely to be detected than scarce proteins. Undersampling is particularly an issue in samples such as plasma or serum that represent the most complex proteome of the human body with proteins spanning 10 to 12 orders of magnitude in linear dynamic range.13 Current mass spectrometers only resolve 4 to 5 orders of magnitude. While a single peptide can be sufficient to unambiguously identify a protein, multiple peptides of the same protein are required for reliable quantification in shotgun proteomics. To date, most proteomics studies analyzing plasma samples have failed to reveal new biomarkers because low-abundant proteins are difficult to detect in the presence of very high-abundant components such as albumin. The issue of undersampling of the plasma proteome could be partially overcome by techniques involving depletion of abundant plasma proteins. An alternative strategy is to use diseased tissue, in which the biomarkers are more enriched. More targeted strategies can be used in plasma/serum once the biomarker candidate has been identified.
Multiple Reaction MonitoringMultiple reaction monitoring on a triple-quadrupole mass spectrometer (QqQ-MS) is applied to peptides or metabolites identified in discovery experiments.14,15 A QqQ-MS provides an effective, accurate way of quantifying a few selected target molecules even in complex mixtures such as plasma or serum.15 If the molecule of interest can be ionized by electrospray ionization, the QqQ-MS will be interfaced to an LC system for separation of the analyte, as mentioned above. The precursor molecule is then selected in the first quadrupole. The second quadrupole is used as a collision chamber for fragmentation. The product ions are detected in the third quadrupole and their intensity is indicative of abundance of the parent molecule.16 Due to its high specificity, multiple reaction monitoring is used for validation of biomarker candidates identified in discovery experiments,17 but it has also been successfully used to confirm specific protein cleavage sites.18 In addition, QqQ-MS is the method of choice for targeted metabolomics studies using standard metabolites as internal calibrators.19
METABOLOMICSMetabolites are the end products of all processes occurring in cells, and metabolite levels in disease reflect the adaptation of biological systems to pathological conditions. It is currently estimated that over 2000 different metabolites can be endogenously synthesized.20 Additionally, exogenous metabolites, such as vitamins, for example, are incorporated as part of the diet. Another major contributor to the metabolite pool is the gut flora. Like proteomics, the aim of metabolomics is to characterize the small molecule complement of a given sample and to interrogate the metabolic networks under normal and pathological conditions in a qualitative and quantitative manner. Metabolomics technologies have been applied to different clinical research areas including biomarkers and drug discovery,21,22 toxicology,23 and nutrition.24 The first metabolomics studies were published early in the 2000s and were rapidly applied to cardiovascular research. As in proteomics, MS is the method of choice for metabolite analyses, but magnetic resonance spectroscopy (MRS) is also widely used.25
Magnetic Resonance Spectroscopy-based MetabolomicsMRS is less sensitive than MS but is highly quantitative and offers advantages for quantifying metabolites in intact tissues and tissue extracts.26 This technique works on the basis that certain nuclei with an odd number of nucleons (protons or neutrons) possess a property called nuclear spin. The resonances of nuclei excited by specific radiofrequencies can be measured to give a distinct signal. Protons (1H) are often the nuclei of choice to measure spin because they are abundant. Using perchloric acid, water-soluble metabolites can be directly extracted from snap-frozen hearts. The 1H-MRS provides a quantitative means of measuring water-soluble metabolites in these metabolite extracts.27 An internal standard is used to calibrate chemical shifts in the spectra for metabolite identifications. Importantly, the peak area corresponding to the signal for each metabolite can be calculated relative to the internal standard in order to obtain quantitative values. Many classes of molecules in the extracted samples can be examined at the same time without prior assumptions as to the molecule types. This makes 1H-NMR an excellent approach for non-targeted analyses. 1H-MRS shows its strength at the required sensitivity mainly in acid extracts. Sensitivity and resolution is diminished in solid tissues. Solid tissues can be analyzed by using a technique called high-resolution magic-angle-spinning 1H-MRS. The energetic state of a given cell or tissue can be assessed by using phosphorus-MRS (31P-MRS). 31P-MRS allows the detection of short-lived cardiac energetic metabolites, such as phosphocreatine and adenosine triphosphate, as well as inorganic phosphate and intracellular pH. 31P-MRS is also well suited for measurements in vivo and in vitro.28,29
Mass Spectrometry-based MetabolomicsMS-based metabolomics provides a highly sensitive means of compiling a metabolic profile, particularly in body fluids.30 Plasma-based metabolomic analyses have been performed after myocardial ischemia,31 in exercise testing, and in patients with prediabetes.32 Metabolites are separated prior to mass spectrometric identification by gas chromatography or LC. As in proteomics, recent progress in MS technologies has transformed our ability to profile metabolites not only in solution but also directly from tissue sections. For example, Manicke et al.33 applied desorption electrospray ionization MS to image and identify lipid species on human plaques. Another study demonstrated small molecule imaging using MS in mouse cardiac tissue.34 We took advantage of the latest MS developments in shotgun lipidomics to compare the lipid content of distinct human atherosclerotic lesions. By comparing endarterectomy samples from symptomatic and asymptomatic patients, and stable and unstable areas within the same symptomatic lesion, we delineated characteristic lipid signatures for plaque vulnerability.18
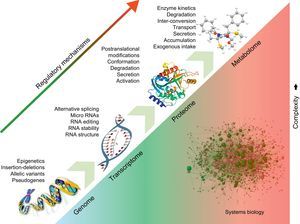
APPLICATIONS TO CARDIOVASCULAR DISEASEUnderstanding Disease MechanismUnlike the genome, the proteome and metabolome are dynamic and are much closer to the disease phenotype (Figure). Thus, they could lead to a better understanding of the processes responsible for the manifestation and progression of CVD. Currently, proteomic techniques can resolve up to 5000 proteins in a complex biological/clinical sample, but proteomics platforms are continuously evolving and push the boundaries of analytical instrumentation.35 In cardiovascular research, the dynamic range in protein expression resulting from abundant myofilament proteins (eg, tropomyosins, myosins, titin) hampers detection of low-abundance proteins in cardiomyocytes. Thus, researchers have developed analytical methods to target specific subproteomes, including cardiac myofilaments,36 cardiac mitochondria,37 cell membranes,38 or nucleus.39 We have recently characterized the remodelling of the vascular extracellular matrix in human abdominal aortic aneurysms, as well as early and late fibrosis in a porcine model of ischemia/reperfusion injury.40,41 By measuring levels of protein expression without a priori assumptions, advanced technologies can help to generate new hypotheses and shift the current focus from quantitative assessment to qualitative differences in cardiac fibrosis that may alter disease progression. The “omics” techniques address the complex interactions within biological systems from a holistic point of view, particularly those interactions involved in the pathophysiology of disease. Previously, conventional reductionist approaches dealt with intracellular signalling cascades as linear models, with the involved molecules confined to single signal pathways. However, different pathways cross-talk with each other and are organized as networks, including proteins as well as small molecules.42 Bioinformatics has become an essential tool to comprehensively analyze and visualize these interactions, and databases such as KEGG (Kyoto Encyclopedia of Genes and Genomes) or Reactome map regulated enzymes/metabolites within different metabolic networks.43 Thus, the combination of proteomics and metabolomics provides a complimentary read-out to improve confidence in data interpretation44 and offers a nonbiased suite of tools to interrogate cardiovascular metabolism.45,46

Systems biology. Regulatory processes at the DNA level affect the expression of downstream molecules, including RNAs, proteins, and metabolites. The effects of the different regulatory elements are additive. Systems biology attempts to analyze the interactions among the different molecular entities in order to offer a holistic view of biological processes and pathological changes occurring in disease.
Atrial fibrillation (AF) leads to several different forms of atrial remodelling, namely electrical, contractile and structural. By using proteomic and metabolomic techniques to analyze the right atrial appendage specimen of patients in sinus rhythm and permanent AF, we revealed a potentially novel form of atrial remodelling-metabolic remodelling.47 Several enzymes involved in glucose, lipid, and energy metabolism were altered during AF. The proteomic findings were substantiated by metabolomic analysis, revealing an increase in lipid metabolites and keton bodies (acetate, beta-hydroxybutyrate). The contribution of metabolic remodelling to AF becoming persistent over time is unknown. However, metabolic alterations appear to precede the onset of AF, suggesting that they are not solely a consequence of AF. In human atrial appendages obtained from patients with and without postoperative AF, dysregulation of cardiac metabolites and altered levels of enzymes relating to energy metabolism occurred before the onset of postoperative arrhythmias.47 Similarly, the observed proteomic and metabolomic changes in an animal model of an evolving AF substrate (ventricular-tachypacing in dogs) point to the central importance of metabolic changes, oxidative stress, and associated structural damage in congestive heart failure and profibrillatory changes in the left atrium.48
Identification of Biomarker CandidatesThe second half of the twentieth century has seen the generalization of minimally-invasive surgical interventions for CVDs, as well as wider application of a variety of pharmacological molecules such as statins, beta-blockers, antiplatelet therapies, and anticoagulants, which have resulted in an enormous improvement in the outcome and prognosis of patients with CVDs. At the same time, the generalization of rapid, noninvasive standard tests to achieve early detection or risk factor assessment have certainly improved diagnosis and preventive therapies. However, CVDs are rarely attributed to a single factor. In the last few years, it has become apparent that there is a need for new biomarkers for CVDs to better stratify patients and determine their response to therapy. Currently, biomarkers are considered as single entities. In the era of high-throughput technologies, new approaches have become available to multiplex biomarkers for clinical diagnosis and prognostication. Mass spectrometers, for example, are already in clinical use for perinatal diagnostics and toxicology. While immunoassays and biochemical methods have dominated clinical analyses, high-throughput techniques could become more widely used for biomarkers of CVD in the forthcoming years.
CONCLUSIONSThe combined use of proteomics and metabolomics, along with new methods for data analysis, permits a more holistic view of biological systems and their alterations in disease. In biological systems, molecules (RNA, protein, or metabolite) interact to execute molecular processes. These complex interactions must not be exclusively addressed by a reductionist approach. The advantage of “omics” techniques is that they can be used for comparisons of clinical samples in a hypothesis-free manner. This has been possible mainly by 3 key factors: a) the improvements achieved in the sensitivity and accuracy of mass spectrometers; b) the development of better separation techniques, together with new labelling methodologies, and c) the availability of databases to analyze datasets of increasing complexity. Mass spectrometers will be the key technology, in proteomics as well as metabolomics, to advance our understanding of CVD and deliver new biomarker candidates.49
CONFLICTS OF INTERESTNone declared.
Prof. Mayr is a Senior Fellow of the British Heart Foundation. The research was supported by the NIHR (National Institute for Health Research) Biomedical Research Centre based at Guy's and St. Thomas’ NHS Foundation Trust and King's College London in partnership with King's College Hospital.