When the first World Health Organization (WHO) meeting on “primary pulmonary hypertension” was held in Geneva in 1973, this entity was considered an orphan disease, and–in the absence of effective medical therapies–this diagnosis was a death sentence for most patients.1 Nowadays, pulmonary hypertension (PH) in general is recognized as a frequent condition, affecting approximately 1% of the global population, and up to 10% of individuals>65 years of age.2 However, most cases of PH occur in association with chronic left heart or lung diseases,2–4 and pulmonary arterial hypertension (PAH)–representing group 1 of the current clinical classification4,5–remains a rather rare entity. The reported prevalence of idiopathic PAH (IPAH), equivalent to “primary pulmonary hypertension” in the early days, is currently between 15 and 26 cases per million population.4,6
During the last 2 decades, tremendous efforts have been undertaken to better understand the pathophysiology of PAH, and the endothelin, nitric oxide, and prostacyclin pathways have been identified as important contributors to disease onset and progression, thus representing meaningful targets for therapeutic interventions.7 As a consequence, numerous targeted PAH therapies were developed, which have been applied as monotherapies or combination therapies.8 In addition to drug development and approval, there has also been a trend in trial design in PAH toward long-term outcome studies using composite morbidity and mortality endpoints.9–11 Recent analyses indicate that aggressive treatment strategies and early combination therapies in particular are associated with improved outcomes.12,13
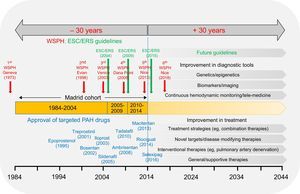
In the article by Quezada Loaiza et al. published in Revista Española de Cardiología, the authors report their experience from a national PH referral center over a period of 30 years.14 In an observational cohort study, the authors describe the clinical approach and prognostic factors of 379 consecutive patients with PAH in whom targeted treatment was initiated between 1984 and 2014. Furthermore, the authors categorized their patients in 3 time periods (1984–2004, 2004–2009, and 2010–2014). The rationale for defining these time periods was based on significant advances in the management of PAH, including the development and availability of targeted medical therapies, and the publication of joint European Society of Cardiology (ESC)/European Respiratory Society (ERS) guidelines for the diagnosis and treatment of pulmonary hypertension in 2004 and 2009.15,16
In 1985, when the first patients had just been included in the present analysis, Robert Zemeckis’ “Back to the future” was released, which brought a whole generation of teenagers into movie theaters. In this movie, Michael J. Fox (alias “Marty McFly”) went on a journey and was transported back 30 years by a time machine. When travelling back in time, McFly was brave enough to use this journey to set directions for the future–saving his mentor (“Doc Brown) from being shot by terrorists, and preventing his father from being permanently bullied by his evil neighbor “Biff”. Unfortunately, we are unable to travel back in time and save PAH patients diagnosed in the early 1980 by applying today's treatment options. However, the data collection of the Madrid PH center enables us to review the clinical impact of advances in the field for patients treated in a large national referral center. The collected data should be put into perspective with various aspects including drug development and emerging treatment concepts, specific recommendations given by several PH world symposia, and the publication of PH guidelines by the ESC/ERS (Figure). In doing so, several factors need to be highlighted:
- 1.
Change of phenotype. It is quite intriguing that the mean age of patients diagnosed with IPAH in the Madrid cohort did not change over time and remained at 44 years, even in the time frame between 2009 and 2014.14 This is in contrast with other registry data such as COMPERA, the UK and French registries, where a constant increase in the median age at diagnosis has been observed in IPAH patients over time, showing a median age of 71, 62 and 55 years, respectively, in the most recent analyses.17–19 In elderly patients, IPAH was often associated with multiple cardiovascular risk factors or comorbidities, which has led to the terms “typical” and “atypical” IPAH.11,20 In the AMBITION study, the latter patients were not eligible for the primary analysis set.11 In general, survival is worse in elderly patients with PAH, despite less impaired pulmonary hemodynamics,17 and the response to targeted therapies may not be the same as in younger patients. The reasons why a stable age range was observed in the Madrid cohort remain speculative.
The authors report a constant presence of IPAH and hereditary PAH, but an increase over time in the number of patients diagnosed with more complex forms of PH including pulmonary veno-occlusive disease (PVOD), congenital heart disease-associated PAH, and portopulmonary hypertension (PoPH).14 The increase of PVOD cases may not reflect a true increase in the incidence of PVOD but may rather be explained by improved knowledge, awareness, and diagnostic tools for this specific entity, including genetic testing.21 Similarly, the relative increase of PoPH is likely explained by the implementation of screening approaches for PoPH, particularly prior to liver tranplant.4
- 2.
Time of diagnosis. Early diagnosis and treatment initiation is considered of key importance in PAH.4 In the Spanish cohort, the authors report that 72% of their patients were in WHO functional class III or IV at the time of diagnosis.14 Unfortunately, this observation is in line with other registries,17–19 indicating that even today most cases of PAH are diagnosed with an unacceptable delay. In fact, the reported mean time from symptom onset to diagnosis in PAH is still 2.8 years. Despite numerous efforts to increase awareness of this disease and improve diagnostic procedures, this has been a rather frustrating experience for PH physicians. Of note, the percentage of PAH patients diagnosed in WHO functional class III or IV in the Madrid center constantly declined from 88.3% before 2004 and 67.1% (2004-2009) to 58% between 2010-2014.14 Hence, this referral center should be congratulated for being able to diagnose PAH patients (and initiate therapy) at an earlier stage over time.
- 3.
Change of therapeutic strategies. Accumulating evidence from numerous recent clinical trials (including but not limited to Pulido et al.,9 Sitbon et al.,10 and Galiè et al.11) has led to substantial changes in the treatment strategies for PAH).4,22 Whereas former recommendations were based on initial monotherapy, followed by sequential escalation based on the concept of treatment goals,15,23 current recommendations are in favor of early or even upfront/initial combination therapy, independently of treatment goals. This is further supported by recent meta-analyses and data from the AMBITION trial (which enrolled only incident patients), suggesting improved outcome of PAH patients with combination therapies.12,13 When looking at registry data, it becomes clear that these concepts are not readily converted into clinical practice.17–20 It is reassuring to note that in the Madrid cohort, the relative number of PAH patients on early combination therapy constantly increased over time from 2.3% before 2004 to 10% between 2004 and 2009, and reached approximately 27% between 2010 and 2014.14 This increase of combination therapies is consistent with current registries, but the implementation of treatment strategies still lags behind guideline recommendations and the generation of clinical evidence. It will be interesting to see whether the frequency of combination therapies further increases in the future in this ongoing observational study.
- 4.
Survival. In their whole cohort, Quezada Loaiza et al. report a survival free from death or transplant for the first, third, and fifth year of 92.2%, 80.6%, and 68.5%, respectively, and the reported median survival was 9 years (95% confidence interval, 7.532–11.959). In the subset of patients with IPAH, the median survival free from death or transplant in the Madrid center was 11 years.14 These numbers are at least comparable to recently published data from the COMPERA, REVEAL, and other registries and highlight the improvements in survival when compared with 2.8 years for patients with primary PH in the National Institutes of Health registry.6,20 This likely reflects the substantial improvements in the field over time, as in general survival improved as treatment options increased. The authors also determined prognostic factors in their cohort and report that survival free from clinical deterioration was higher in women, patients younger than 56 years, patients with right atrial pressure<8mmHg, with 6-minute walk distance>475 meters, and those in WHO functional class I-II. This is consistent with the risk stratification for PAH patients recommended in the current ESC/ERS guidelines.4 The particularly favorable survival rate in the Madrid cohort may be explained by the fact that patients were rather young at diagnosis, had better WHO functional class, and achieved a longer 6-minute walk distance when compared with patients in other registries.
- 5.
Toxic rapeseed oil syndrome epidemic. A unique aspect of the Spanish cohort is the toxic rapeseed oil syndrome epidemic that occurred in Spain in the early 1980s. Quezada Loaiza et al. report that during this epidemic approximately 4000 affected individuals developed PAH, which was however spontaneously reversible in the vast majority of cases.14 Nevertheless, a small subset developed chronic severe PAH over time, with wide variations in latency. While approximately 75% of these patients were diagnosed prior to 2004, representing 20.3% of PAH patients between 1984 and 2004, such cases continued to be diagnosed in the other time periods, representing 2.9% (2004-2009) and 4.9% (2010-2014) of newly diagnosed PAH patients, thus indicating a highly variable latency. This is consistent with the concept that the onset and progression of chronic PAH is not induced by a single factor, but requires a “second hit” and/or the combination of a trigger and genetic susceptibility in individual patients.
 such as World Symposia on Pulmonary Hypertension (WSPH; red), the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) guidelines on pulmonary hypertension (green), and the introduction of targeted pulmonary arterial hypertension therapies (blue). In addition, future developments (+ 30 years) are outlined, which include improvement of diagnostic tools and treatment strategies, and future guidelines. PAH, pulmonary arterial hypertension.")
Back to the future in pulmonary arterial hypertension. Data collected from the Madrid cohort14 were put into perspective to previous developments ( 30 years) such as World Symposia on Pulmonary Hypertension (WSPH; red), the European Society of Cardiology (ESC) and the European Respiratory Society (ERS) guidelines on pulmonary hypertension (green), and the introduction of targeted pulmonary arterial hypertension therapies (blue). In addition, future developments (+ 30 years) are outlined, which include improvement of diagnostic tools and treatment strategies, and future guidelines. PAH, pulmonary arterial hypertension.
What could the future bring us? Improved understanding of genetics/epigenetics, the establishment of novel biomarkers, as well as improved imaging techniques should help us to detect PAH at earlier stages, and to better capture high-risk scenarios and treatment responses in both the pulmonary circulation and right ventricle. It is important to note that–despite significant improvements–we remain far from being able to cure PAH, and patients are still affected by significant morbidity and mortality. Hence, it is crucial to identify further treatable targets and to develop additional, disease-modifying therapies. Nowadays, PAH is not only considered to result from a dysregulation of vascular tone, but is recognized as a proliferative disease of the small pulmonary resistance vessels. Current research approaches focus on antiremodeling strategies, inflammatory mediators, and mitochondrial function and metabolism.7,24 Is this new? PAH was probably first described in 1865 by Julius Klob, a pathologist from Austria, who described “endarteriitis pulmonalis deformans” as a “disease that is characterized by an increase in mass of the inner vessel skin which grows out to form a pseudomembraneous connective tissue”.25 Astonishingly and intriguingly, Klob also noted that “this entity was occasionally–but not always–associated with inflammation”. Thus, what was observed more than 150 years ago still appears to be the basis for today's modern research concepts. The first clinical step toward antiremodeling therapies may have been the IMPRES trial, which demonstrated that the tyrosine kinase inhibitor imatinib–acting via antiproliferative and proapoptotic actions–substantially improved exercise capacity and pulmonary hemodynamics in severe PAH, when given in addition to approved targeted therapies.26 While safety concerns impeded the approval of imatinib for PAH, it may be of key importance to follow-up on this promising strategy and to further develop therapies that truly target the underlying morphological alterations of the pulmonary arterioles.
In addition to modern pharmacotherapy, general measures and supportive therapies are also considered important.4 For instance, exercise training and rehabilitation programs have proven safe and effective in patients with PAH, and were included in current guideline recommendations with a high level of evidence.4 However, structured training programs are sparse, and the widespread applicability and reliable continuation of exercise beyond the initial training phase remain important challenges. Recently, whole body vibration, which is performed on a vibrating platform that moves in sinusoidal oscillations, was introduced as a novel exercise modality and was shown in a controlled pilot study to improve exercise capacity, physical performance, and health-related quality of life within 4 weeks.27 Potentially, whole body vibration may be included in future structured training and rehabilitation concepts and may provide an opportunity for continuous long-term, home-based physical exercise in these patients.
Another parallel with the initiation of the Madrid study–although darker than Zemeckis’ movie–is George Orwell's “1984”. In that novel, originally written in 1948, Orwell (“big brother is watching you”) tried to project contemporary social developments 36 years into the future–in this case resulting in a dark picture of permanent and inescapable surveillance by totalitarian structures. In modern medicine, remote data monitoring and health status observation may be viewed as beneficial and open a wide variety of applications for the future, such as the ability to detect disease deterioration at earlier time points. For instance, remote monitoring of pulmonary artery pressure was recently shown to substantially reduce the rate of hospitalizations among heart failure patients in the CHAMPION trial.28 However, there may also be a scary side to devices that are able to track and automatically transmit individual health/activity status or geographical positions to “a database”. Indeed, it will be of key importance to develop such promising telemonitoring tools in the most responsible way.
Finally, an important future task will be to look beyond PAH and better understand the pathophysiology and establish therapies for other forms of PH that are much more common and have a strong impact on survival. Given the demographic development in aging populations, the increase in prevalence of heart failure with preserved ejection fraction (HFpEF), and the high prevalence of PH in HFpEF, this is particularly true of PH-HFpEF. While the primary disease to target is clearly located in the left heart, the pulmonary circulation likely represents an additional treatment target at least in a subset of patients.3 However, future research in this field will require detailed phenotyping, including precise hemodynamic characterization at rest, improvement of our understanding of hemodynamic changes during exercise, and controlled outcome studies in well-defined patient populations, some of which are underway.29
Since the completion of data acquisition of the current Madrid cohort, the 2015 ESC/ERS guidelines for the diagnosis and treatment of PH have been published,4 and in 2018, recent advances in the field will be reviewed, and new recommendations made at the sixth World Symposium on Pulmonary Hypertension in Nice, France. It will be interesting to see how these new recommendations and future developments will impact patient management and outcome in the ongoing observation of the Madrid referral center. The future has already begun.
CONFLICTS OF INTERESTS. Rosenkranz has received remunerations for lectures and/or consultancy from Actelion, Bayer, GSK, MSD, Novartis, Pfizer and United Therapeutics. His institution has received research grants from Actelion, Bayer, MSD, Novartis, and United Therapeutics. D. Dumitrescu has received remunerations for lectures and/or consultancy from Actelion, Bayer, GSK, Novartis and Servier.