Type A aortic dissection is a condition associated with high mortality. Early diagnosis and surgical treatment are essential because of the risk of rapidly developing serious complications such as severe aortic regurgitation, cardiac tamponade, aortic rupture, and even death in a high percentage of cases.
A variety of risk factors are known to contribute to aortic rigidity, favoring the development of both aneurysms and dissections. Of these factors, hypertension is one of the most important.1 Fibrous intimal thickening and increased proteoglycans and CD68-positive macrophages/histiocytes are usually observed in aortic aging. These findings are associated with atherosclerosis.1 Several genetic syndromes are also associated with ascending aortic aneurysms and dissections. The most frequent and well known are Marfan syndrome and bicuspid aortic valve, but others such as Loeys-Dietz syndrome and Turner syndrome are also relatively frequent causes. These syndromes share certain histopathological characteristics, mainly cystic medial degeneration.1 An abnormal finding common to these syndromes is upregulation of the activity of transforming growth factor β in the ascending aorta.2
More recently, an entity has been described known as familial aneurysm and dissection. This entity has few evident physical characteristics and has been associated with mutations in multiple genes, including smooth muscle α-actin, transforming growth factor β, and myosin heavy chain 11.3 The following criteria need to be met for diagnosis: type A or type B aortic dilatation or dissection in individuals younger than 60 years, in the absence of other connective tissue syndromes, hypertension, or atherosclerotic disease and pathological evidence of cystic necrosis of the aortic media or a positive family history (aneurysm or aortic dissection, unexplained sudden death, or known genetic mutation).3
Up to 40% of thoracic aortic dissections are expected to be hereditary, and therefore study of a specific underlying genotype may help confirm diagnosis and enable appropriate monitoring, individualized medical and surgical treatment, and screening of other family members to reduce morbidity and mortality.4 This prompted the American College of Cardiology Foundation and the American Heart Association to recommend that the underlying genetic mutation should determine the timing of aortic repair.5
Therefore, given the implications of identifying these entities for other family members, it is essential to reach an etiologic diagnosis. But do we always reach one?
To address this question, we designed a study with the objective of assessing how many final etiologic diagnoses were made among patients with type A dissection in our center between 2000 and 2016.
This was a retrospective study in which all patients diagnosed with type A dissection in the study period were assessed. We reviewed the medical histories of the deceased and recorded whether or not an autopsy had been performed. Among survivors, we also recorded whether final a etiologic diagnosis was reached.
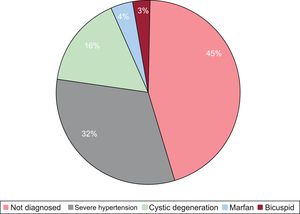
Information from 75 patients was analyzed. Of these patients, 47 were male (63%), the mean age at diagnosis was 63.09±13.8 years, and the mean duration of follow-up was 76±49 months. The following risk factors were present: hypertension (53.3%), diabetes mellitus (8%), dyslipidemia (15%), and smoking (12%). The intraoperative mortality rate was 6.7% (5 patients) and the in-hospital mortality rate was 25.3% (19 patients). Autopsy was performed in 5 of the patients who died (6.7%). During follow-up, 6 patients (8%) died and 11 patients (14.7%) were lost to follow-up. Two of the patients had a relevant family history and 4 (5.3%) had undergone genetic study. The etiologic diagnosis yielded the following results: Marfan syndrome in 3 patients (4%), bicuspid valve in 2 (2.7%), cystic degeneration in 12 (16%), and severe hypertension in 24 (31.8%). Definitive etiologic diagnosis was not established in 34 patients (45.2%), although aortic wall degeneration was reported in 16 patients (21.2%) (Figure).

It was therefore concluded that etiologic diagnosis was not reached in a significant number of patients with type A dissection even though this is essential information for family counselling. While syndromic entities are readily diagnosed, this is not the case for entities such as familial aneurysm and dissection that require family screening and that are associated with cystic medial degeneration. Therefore, the family history and the histologic study of the diseased aortic wall, as well as genetic study, if available, are key in this disease. Likewise, it is important that patients diagnosed with aortic dissection are managed by multidisciplinary teams able to improve morbidity and mortality both of patients and, in some cases, their family members.