According to sudden cardiac death guidelines, an implantable cardioverter-defibrillator (ICD) should be considered in patients with LMNA-related dilated cardiomyopathy (DCM) and ≥ 2 risk factors: male sex, left ventricular ejection fraction (LVEF) <45%, nonsustained ventricular tachycardia (NSVT), and nonmissense genetic variants. In this study we aimed to describe the clinical characteristics of carriers of LMNA genetic variants among individuals from a Spanish cardiac-laminopathies cohort (REDLAMINA registry) and to assess previously reported risk criteria.
MethodsThe relationship between risk factors and cardiovascular events was evaluated in a cohort of 140 carriers (age ≥ 16 years) of pathogenic LMNA variants (54 probands, 86 relatives). We considered: a) major arrhythmic events (MAE) if there was appropriate ICD discharge or sudden cardiac death; b) heart failure death if there was heart transplant or death due to heart failure.
ResultsWe identified 11 novel and 21 previously reported LMNA-related DCM variants. LVEF <45% (P=.001) and NSVT (P <.001) were related to MAE, but not sex or type of genetic variant. The only factor independently related to heart failure death was LVEF <45% (P <.001).
ConclusionsIn the REDLAMINA registry cohort, the only predictors independently associated with MAE were NSVT and LVEF <45%. Therefore, female carriers of missense variants with either NSVT or LVEF <45% should not be considered a low-risk group. It is important to individualize risk stratification in carriers of LMNA missense variants, because not all have the same prognosis.
Keywords
Genetic variants in the lamin gene (LMNA) cause between 5% and 10% of dilated cardiomyopathies (DCMs). LMNA-related DCM is associated with conduction disorders, atrial and ventricular arrhythmias, a high incidence of premature sudden cardiac death (SCD), and progression to end-stage heart failure.1–5
American and European guidelines on SCD recommend an implantable cardioverter-defibrillator (ICD) for patients with 2 or more risk factors: nonsustained ventricular tachycardia (NSVT), left ventricular ejection fraction (LVEF) < 45% at the initial evaluation, male sex, and different missense mutations (insertions, deletions, truncations, or mutations affecting splicing).6,7 A new arrhythmic risk score based on previous risk factors has recently been proposed; it considers LVEF a continuous variable and includes a new factor: first-degree or higher atrioventricular block (AVB). This score indicates an ICD if the 5-year SCD risk is ≥ 7%.8 According to both stratification methods, an ICD would not be recommended in a woman with DCM who has an LMNA missense variant and NSVT or LVEF < 45%.
The objectives of this study were to a) clinically characterize the population of patients with LMNA variant-related DCM and their families (healthy and affected carriers); b) describe new LMNA variants and possible genotype-phenotype correlations; and c) reevaluate arrhythmic risk factors previously described in our cohort.
METHODSData were retrospectively collected on patients with LMNA-related DCM who were being followed up in 18 Spanish cardiology departments as part of the Spanish Registry of Cardiac Laminopathies in Adults (REDLAMINA registry). The registry includes carriers ≥ 16 years old at the first cardiac evaluation. Cardiac laminopathy was defined as the presence of a pathogenic or likely pathogenic genetic variant in the LMNA gene in any patient with a predominantly cardiac phenotype (DCM or hypokinetic nondilated cardiomyopathy), conduction disorder, ventricular or supraventricular arrhythmia, or premature SCD. All relatives identified in the family screening as being carriers of a pathogenic LMNA variant were included in the analysis, independently of their clinical status (healthy or not). Data were collected at the first cardiology visit and the last follow-up. These data included personal and family medical histories, results of genetic studies, functional class, and relevant echocardiographic data, electrocardiogram results, Holter monitoring findings, and magnetic resonance images. The anonymized information was collected and analyzed in the University Hospital Complex of A Coruña, Spain. The study was approved by the local ethics committee (registry number, 2017/300). The complete methods can be consulted in the .
Genetic studies were performed in each participating center in accordance with local protocols, and the recruitment period lasted from 1999 to 2018. Genetic variants were divided into missense and nonmissense (insertion, deletion, truncation, or mutations affecting splicing).
Recommendations of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology were followed to determine the pathogenicity of the identified variants.9 Two cardiologists with experience in genetic variation interpretation agreed on the final classification of each variant. Variants considered to be probably benign, nonpathogenic, or of unknown significance were excluded from the study.
Also excluded were genetic variants not related to predominantly cardiac phenotypes (eg, lipodystrophy, metabolic syndrome, polyneuropathies) ( and ).
Statistical analysisContinuous variables are expressed as mean±standard deviation and were compared using either the t test or Mann-Whitney U test according to their distribution. Noncontinuous variables are expressed as absolute and relative frequencies and were compared using the chi-square or Fisher exact test, as appropriate. P < .05 was considered significant. The analysis was performed using R software, version 3.4.3 (R Foundation for Statistical Computing).
The following composite end points were analyzed: a) major arrhythmic events (MAEs), if appropriate ICD discharge or SCD was recorded, and b) heart failure death (HFD), if a heart transplant (HTx) was performed or death occurred due to heart failure.
The cumulative probability of MAEs and HFD was calculated using the Kaplan-Meier method. Factors were compared using the Mantel-Cox log-rank test. The initial date of diagnosis was taken as the start of follow-up. Cox regression analysis was used to identify independent predictors of events. Risk ratios and 95% confidence intervals (95%CIs) for end points were calculated with Cox proportional hazards models, which included as covariables sex, type of genetic variant (nonmissense vs missense), LVEF < 45%, first-degree AVB, complete AVB, or any degree of AVB with NSVT.
RESULTSData were collected on 222 LMNA variant carriers. Six patients from 1 family were excluded due to lack of follow-up. Sixty carriers were excluded because their variants were considered nonpathogenic or their phenotype was different to that defined as cardiac laminopathy, as well as 16 carriers that were younger than 16 years old at the initial evaluation. In total, 140 carriers (54 probands and 86 relatives) were included in the final analysis (). The median [interquartile range] duration of follow-up was 5.0 [2.0-9.25] years for the probands and 3.0 [1.0-6.0] years for family members.
Genetic variants identified in the REDLAMINA registryIn the REDLAMINA registry cohort, 11 new pathogenic variants were identified (4 missense and 7 nonmissense), as well as 21 known variants (16 missense and 5 nonmissense) (). The most frequent variant was p.Arg190Trp, described in 16 carriers from 5 different families.
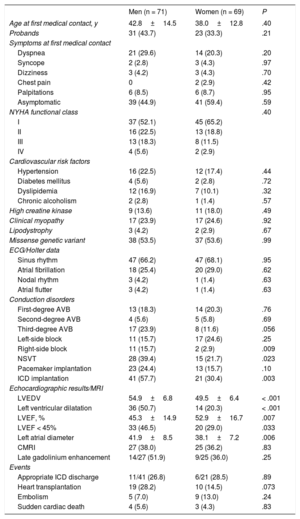
Clinical characteristicsThe clinical characteristics of the cohort are summarized in table 1, and more detail can be found in . At first medical contact, no significant differences were found between men and women in age, symptoms, New York Heart Association (NYHA) functional class, clinical myopathy, or coronary risk factors. Left ventricular dilatation and systolic dysfunction were more common in men. There was a high incidence of conduction disorders, with 42.9% of carriers having some degree of AVB and 17.9% having third-degree AVB, which was nonsignificantly more frequent in men (P = .057).
Differences in characteristics and clinical events between men and women with LMNA variants in the REDLAMINA registry cohort
| Men (n = 71) | Women (n = 69) | P | |
|---|---|---|---|
| Age at first medical contact, y | 42.8±14.5 | 38.0±12.8 | .40 |
| Probands | 31 (43.7) | 23 (33.3) | .21 |
| Symptoms at first medical contact | |||
| Dyspnea | 21 (29.6) | 14 (20.3) | .20 |
| Syncope | 2 (2.8) | 3 (4.3) | .97 |
| Dizziness | 3 (4.2) | 3 (4.3) | .70 |
| Chest pain | 0 | 2 (2.9) | .42 |
| Palpitations | 6 (8.5) | 6 (8.7) | .95 |
| Asymptomatic | 39 (44.9) | 41 (59.4) | .59 |
| NYHA functional class | .40 | ||
| I | 37 (52.1) | 45 (65.2) | |
| II | 16 (22.5) | 13 (18.8) | |
| III | 13 (18.3) | 8 (11.5) | |
| IV | 4 (5.6) | 2 (2.9) | |
| Cardiovascular risk factors | |||
| Hypertension | 16 (22.5) | 12 (17.4) | .44 |
| Diabetes mellitus | 4 (5.6) | 2 (2.8) | .72 |
| Dyslipidemia | 12 (16.9) | 7 (10.1) | .32 |
| Chronic alcoholism | 2 (2.8) | 1 (1.4) | .57 |
| High creatine kinase | 9 (13.6) | 11 (18.0) | .49 |
| Clinical myopathy | 17 (23.9) | 17 (24.6) | .92 |
| Lipodystrophy | 3 (4.2) | 2 (2.9) | .67 |
| Missense genetic variant | 38 (53.5) | 37 (53.6) | .99 |
| ECG/Holter data | |||
| Sinus rhythm | 47 (66.2) | 47 (68.1) | .95 |
| Atrial fibrillation | 18 (25.4) | 20 (29.0) | .62 |
| Nodal rhythm | 3 (4.2) | 1 (1.4) | .63 |
| Atrial flutter | 3 (4.2) | 1 (1.4) | .63 |
| Conduction disorders | |||
| First-degree AVB | 13 (18.3) | 14 (20.3) | .76 |
| Second-degree AVB | 4 (5.6) | 5 (5.8) | .69 |
| Third-degree AVB | 17 (23.9) | 8 (11.6) | .056 |
| Left-side block | 11 (15.7) | 17 (24.6) | .25 |
| Right-side block | 11 (15.7) | 2 (2.9) | .009 |
| NSVT | 28 (39.4) | 15 (21.7) | .023 |
| Pacemaker implantation | 23 (24.4) | 13 (15.7) | .10 |
| ICD implantation | 41 (57.7) | 21 (30.4) | .003 |
| Echocardiographic results/MRI | |||
| LVEDV | 54.9±6.8 | 49.5±6.4 | < .001 |
| Left ventricular dilatation | 36 (50.7) | 14 (20.3) | < .001 |
| LVEF, % | 45.3±14.9 | 52.9±16.7 | .007 |
| LVEF < 45% | 33 (46.5) | 20 (29.0) | .033 |
| Left atrial diameter | 41.9±8.5 | 38.1±7.2 | .006 |
| CMRI | 27 (38.0) | 25 (36.2) | .83 |
| Late gadolinium enhancement | 14/27 (51.9) | 9/25 (36.0) | .25 |
| Events | |||
| Appropriate ICD discharge | 11/41 (26.8) | 6/21 (28.5) | .89 |
| Heart transplantation | 19 (28.2) | 10 (14.5) | .073 |
| Embolism | 5 (7.0) | 9 (13.0) | .24 |
| Sudden cardiac death | 4 (5.6) | 3 (4.3) | .83 |
AVB, atrioventricular block; CMRI, cardiac magnetic resonance imaging; ECG, electrocardiogram; ICD, implantable cardioverter-defibrillator; LVEDV, left ventricular end-diastolic volume; LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia; NYHA, New York Heart Association.
Values express No. (%) or median±standard deviation.
ICDs were implanted in 62 carriers, with a higher frequency in men (41 of 71; 58%) than in women (21 of 69; 30%) (P = .003). Of the total devices implanted, 90% (56 carriers: 20 women and 36 men) were in primary prevention and 10% (6 carriers: 5 men and 1 woman) were in secondary prevention.
EventsAt the end of follow-up, 7 SCDs were documented, 4 in probands and 3 in relatives (detected during the family study). Five SCDs occurred in the 78 carriers that did not undergo device implantation ().
There was only 1 death due to advanced heart failure (1 proband with contraindications for HTx). More HTxs were performed in probands (17 of 52; 32.7%) than in family members (12 of 88; 13.6%) (P = .007). In the subgroup of carriers that received an ICD, the proportion of appropriate discharges was similar in probands and family members (11 of 36 [27.9%] vs 6 of 26 [22.2%]; P = .6).
There was no significant difference in the frequency of events (embolic events, SCD, ICD discharges, or HTx) between men and women (table 1).
Low-risk carriers according to guidelines and Wahbi scoreTwo low-risk carriers who had experienced SCD were detected (both carriers of the p.Arg190Trp variant). The guidelines and Wahbi score were used to identify the remaining carriers who experienced SCD or who had an appropriate ICD discharge6–8 ().
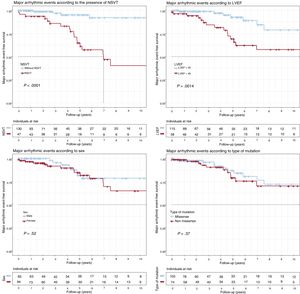
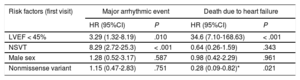
Survival analysisMajor adverse events (figure 1)LVEF < 45% at the start of follow-up (P = .001) and NSVT (P < .0001) were associated with a worse survival rate. Both factors were independently associated with MAEs in the Cox regression model, with hazard ratios (HRs) of 3.29 (95%CI, 1.32-8.19) and 8.29 (95%CI, 2.72-25.3), respectively (table 2). No significant differences were identified in the type of genetic variant (P = .37), sex (P = .52), complete AVB (P = .55), or any degree of AVB (P = .32).
. Composite end point: major arrhythmic events (appropriate ICD discharge/sudden cardiac death) during follow-up. ICD, implantable cardioverter-defibrillator; LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia.")
Kaplan-Meier survival analysis for NSVT, LVEF < 45%, sex, and type of genetic variant (missense vs nonmissense). Composite end point: major arrhythmic events (appropriate ICD discharge/sudden cardiac death) during follow-up. ICD, implantable cardioverter-defibrillator; LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia.
Evaluation of classic risk factors for different composite events in the LMNA genetic variant carriers of the REDLAMINA registry cohort (Cox regression model)
| Risk factors (first visit) | Major arrhythmic event | Death due to heart failure | ||
|---|---|---|---|---|
| HR (95%CI) | P | HR (95%CI) | P | |
| LVEF < 45% | 3.29 (1.32-8.19) | .010 | 34.6 (7.10-168.63) | < .001 |
| NSVT | 8.29 (2.72-25.3) | < .001 | 0.64 (0.26-1.59) | .343 |
| Male sex | 1.28 (0.52-3.17) | .587 | 0.98 (0.42-2.29) | .961 |
| Nonmissense variant | 1.15 (0.47-2.83) | .751 | 0.28 (0.09-0.82)* | .021 |
95%CI, 95% confidence interval; HR, hazard ratio; LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia.
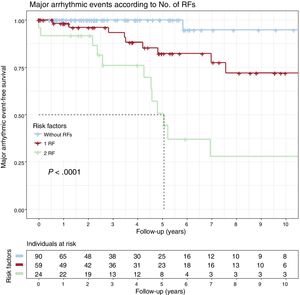
The cumulative MAE-free survival rates for carriers with 0, 1 (NSVT or LVEF < 45%), or 2 (NSVT and LVEF < 45%) risk factors can be seen in figure 2. Only 1 carrier without risk factors had an MAE; the SCD-free survival rate at the 5-year follow-up was 82.3% with only 1 risk factor and 50.7% with 2 (P < .001).
Heart failure death (figure 3)
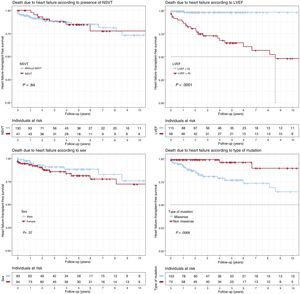
Patients with LVEF < 45% at first medical contact showed significantly shorter survival (P < .001), as did missense variant carriers (P = .007). Only LVEF < 45% was independently associated with heart failure death or HTx in the Cox regression model (HR = 25.1; 95%CI, 5.76-109.18). There were no significant differences according to sex (P = .37) or the presence of NSVT (P = .84).
. Composite end point: heart transplantation or death due to heart failure during follow-up. LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia.")
Kaplan-Meier survival analysis for NSVT, LVEF < 45%, sex, and type of genetic variant (missense vs nonmissense). Composite end point: heart transplantation or death due to heart failure during follow-up. LVEF, left ventricular ejection fraction; NSVT, nonsustained ventricular tachycardia.
Of the 16 carriers described, 7 had HTxs and 2 experienced SCD. An asymptomatic 28-year-old man with slight left ventricular dilatation and normal LVEF experienced SCD while sleeping. He had normal Holter and stress test findings. His mother was also a p.Arg190Trp carrier and was diagnosed with DCM and 40% LVEF at 43 years of age; her atrial fibrillation had reverted to sinus rhythm and she had no NSVT on Holter monitoring. She died suddenly while in NYHA class II and under medical treatment.
DISCUSSIONOur series, which evaluates a Spanish cohort of patients with cardiac laminopathies and related carriers, provides new information on the clinical relevance and prognosis of this condition. The clinical characteristics of 140 carriers with 32 DCM-related LMNA variants (11 new and 21 previously described) are described in detail, contributing to existing knowledge on genotype-phenotype correlations (). Current risk stratification criteria were also reevaluated. In the REDLAMINA registry cohort:
- •
Women showed the same risk of MAEs and DCM as men.
- •
LVEF < 45% was the only factor independently related to DCM, whereas sex and NSVT were not significant predictors.
- •
No differences in MAEs were identified according to type of variant (nonmissense vs missense) but our results did show that some missense variants might share the poor prognosis of nonmissense variants.
American and European guidelines recommend an ICD in patients with 2 or more of the following risk factors: LVEF < 45%, NSVT, nonmissense variants, or male sex. In their new scale, Wahbi et al.6–8 added first-degree or higher AVB and LVEF as a linear variable. These recommendations are mainly based on a 2012 article by van Rijsingen et al.10 that included a European cohort of 6 countries comprising 109 families and a total of 269 carriers. Wahbi et al.8 also used data from this cohort.
Relationship between the type of LMNA genetic variant and prognosisAccording to current recommendations, the presence of a single LMNA nonmissense variant is associated with a worse prognosis than a missense variant.6–8 However, this difference is not confirmed by our study.
One possible reason is the pathogenicity attributed to some variants included in the original study by van Rijsingen et al.10 With pathogenicity as a criterion, the variants collected in that cohort were absent in at least 150 ethnically compatible controls. However, of the 37 missense genetic variants included in the original article, at least 6 (p.Lys117Arg, p.Arg397Cys, p.Arg545His, p .Ser573Leu, p.Gly638Arg, and p.Arg644Cys) can currently be classified as probably nonpathogenic or of unknown significance (). All of these variants have low but significant frequencies in public databases, such as gnomAD,11 and are classified as either probably nonpathogenic or of unknown significance in ClinVar.12 With the information available in 2012, it was impossible to perform this evaluation. These variants were found in 12 index cases and 19 family members that should have been excluded from the analysis. The inclusion of nonpathogenic LMNA variants and, as a result, the inclusion of DCM of other etiologies that may have had a more benign clinical course, could explain the different prognosis observed in missense genetic variant carriers.
In the Wahbi et al. score, the authors do not mention the genetic variants included in the study. It is therefore impossible to confirm their pathogenicity. Their work also included patients with phenotypes other that of cardiac laminopathy (eg, 72 patients with lipodystrophy, 65 with Emery-Dreifuss), which may have influenced the results.8,13
The REDLAMINA registry included only pathogenic or genetic variants with a high probability of being pathogenic (according to present criteria),9 excluding variants not clearly related to the cardiac laminopathy phenotype, as defined in the Methods section ( and ).
The other possible explanation for the discrepancy in the results is that not all missense pathogenic variants included in these studies have the same prognosis. The missense genetic variants included in the REDLAMINA registry cohort might be associated with a worse prognosis than those included in both the European and Wahbi et al.8 cohorts.
Differences between men and womenIn the present registry, the risk of MAEs and DCM was similar in men and women. However, the number of ICDs implanted was higher in men than in women (table 1). Although this could be considered to be due to the strict application of the guidelines (given that male sex is a risk factor), every center in the REDLAMINA registry applied its own criteria when implanting an ICD (). This approach is not uncommon for patients with rare diseases associated with SCD.
In our cohort, men had a lower LVEF at first examination and, even though the number of HTxs was higher in men than in women, there were no significant differences between the sexes in the incidence of this event (SCD) during follow-up (figure 3). Neither were there differences between men and women in final composite events (MAEs and DCM) or in embolic phenomena such as SCD, AVB, pacemaker implantation, or ICD discharges (table 1).
The results of previous cohorts could have been influenced by the inclusion of nonpathogenic missense variants in previous studies and the different prognoses of some missense variants.10,14
“Low-risk” LMNA missense variants“Low-risk” missense variants with the founder effect have been described in the literature. As well as the p.Arg331Gln (58 carriers) and p.Arg216Cys (36 carriers) variants, they have been associated with a delayed presentation and a good prognosis vs other pathogenic LMNA variants.15,16 Captur et al.17,18 carried out a study of all LMNA genetic variants published and their relationship with the phenotype described and found that malignant ventricular arrhythmias occur with greater frequency in nonmissense variant carriers. Interestingly, they observed that not all missense variants confer the same prognosis.
“High-risk” LMNA missense variantsIn contrast, some LMNA missense variants are associated with poor prognosis. A clear example is the most frequent variant reported in the REDLAMINA registry: p.Arg190Trp. The REDLAMINA registry describes 2 carriers of this missense variant. According to the established risk criteria (the guidelines and the Wahbi et al. score8), these patients did not require ICD implantation but nevertheless experienced SCD. Both carriers had only 1 risk factor (the male patient because of his sex and the female patient because her LVEF was reduced). Application of the Wahbi score would also not have indicated the need for ICD implantation (< 7%). However, a review of the literature concerning this variant would probably have led to a “high-risk” classification. At least 19 articles provide clinical information on 23 affected families, with a high incidence of HTx, SCD, and heart failure. This variant was mentioned for the first time in 2002 by Arbustini et al.19 in an Italian family with DCM and SCD. It was subsequently described on various occasions in other European, Asian, and American families and always associated with unfavorable prognoses ().
Current risk criteria underestimate the risk of missense variantsOur data indicate that the current risk criteria in cardiac laminopathies are not appropriate for missense variant carriers because not all of them have the same prognosis. American and European guidelines, as well as the Wahbi et al. score, underestimate the risk of these missense variants.6–8 The work of van Rijsingen et al.10,14 and Wahbi et al.,8 apart from the already mentioned limitations, do not provide clinical data that allow us to determine which events are related to which particular variant.13 It is essential to develop international registries that share clinical information on LMNA missense variants that would allow the deduction of prognostic information and in turn help to stratify SCD risk.
The current recommendations concerning ICD implantations in cardiac laminopathies represent a good strategy but can underestimate the risk of SCD in women with certain missense variants. To manage these aspects, more studies overcoming the above limitations are needed.
LimitationsThe REDLAMINA registry has all of the limitations inherent to a retrospective multicenter study. Because most participating hospitals are referral centers with HTx programs, there could be a selection bias regarding the severity of the patients included in the study. The REDLAMINA registry compiled data from various centers, which may have introduced an unforeseen bias. Our sample size is smaller than that of previously published registries, although it has the advantage of being from a single country, in contrast to others that might be biased by the use of different patient treatments. The most frequent variant was p.Arg190Trp (already associated with an unfavorable prognosis), which might have influenced the results.
In our cohort, few events occurred in the low-risk groups, and no definitive conclusions can be drawn, which could be a limitation of our study. In fact, only 17 MAEs were recorded, lower than of the other mentioned series. Nevertheless, it must be considered that these series also included events related to missense variants with very doubtful pathogenicity (such as in the series of van Rinjensen et al.10,14) or simply did not provide data on which missense variants were related to events, as in the Wahbi et al. series.8 In addition, in the REDLAMINA registry, when “high-risk” patients (with at least 2 risk factors) were considered, there were no significant differences between men and women, as previously demonstrated.
Not all patients included in our study underwent complete screening (through massive ultrasequencing) for genes related to DCM because some of the older studies were performed using the Sanger technique. This could signify a bias, given that the patients whose LMNA gene was exclusively studied could have a pathogenic variant in another gene that may have influenced the prognosis.
Another limitation could also be the low number of patients that underwent cardiac magnetic resonance imaging (52 patients out of 140 included).
CONCLUSIONSIn the REDLAMINA registry cohort, the only 2 independent predictors associated with MAEs were NSVT and LVEF < 45% and not sex and type of genetic variant (missense vs nonmissense). Therefore, people with missense variants with NSVT or LVEF < 45% should not be considered a low-risk group. For an appropriate risk stratification, the prognosis of LMNA missense variants needs to be individualized, given that some could have as poor a prognosis as nonmissense variants.
FUNDINGThis study received a grant from the Proyecto de investigación de la Sección de Insuficiencia Cardiaca 2017 from the Spanish Society of Cardiology and grants from the Instituto de Salud Carlos III (ISCIII) [PI14/0967, PI15/01551, AC16/0014] and ERA-CVD Joint Transnational Call 2016 (Genprovic). Grants from the ISCIII and the Ministerio de Economía y Competitividad de España (Spanish Department of Economy and Competitiveness) are supported by the Plan Estatal de I+D+i 2013-2016: Fondo Europeo de Desarrollo Regional (FEDER) “Una forma de hacer Europa”. The Hospital Universitario Puerta de Hierro Majadahonda and the Hospital Virgen de la Arrixaca are members of the European Reference Network for rare and complicated heart diseases (ERN GUARD-Heart). The funding bodies were not involved in the design, collection, analysis, or interpretation of the data nor the decision to send the manuscript for publication.
- –
Genetic variants in the lamin gene (LMNA) cause between 5% and 10% of dilated cardiomyopathies (DCMs) and are associated with conduction disorders, arrhythmias, premature SCD, heart failure, and HTx. According to the guidelines, an ICD should be considered when patients have 2 or more of the following risk factors: male sex, LVEF < 45%, NSVT, and nonmissense variants.
- –
The characteristics of a Spanish cohort of 140 LMNA variant carriers (≥ 16 years) are described and current risk factors evaluated. The only 2 predictors associated with SCD or appropriate ICD discharge were NSVT and LVEF < 45%; LVEF < 45% was associated with DCM. No associations were found for sex or genetic variant.
- –
Our data indicate that current criteria underestimate the risk of missense variants because not all share the same prognosis. People with missense variants with NSVT or LVEF < 45% should not be considered low risk and it is important to assess the risk individually.
L. Montserrat is the CEO of Health in Code S.L. None of the other authors have any conflicts of interest to declare.
To Cristina Martinez-Veira, Diana Castro-Dios, Efrén Freije-Botana, and David Martos for their invaluable help with the REDLAMINA registry.
Supplementary data associated with this article can be found in the online version available at https://doi.org/10.1016/j.rec.2020.03.026
The names of all of the researchers and centers participating in the REDLAMINA registry are listed in the