Pulmonary hypertension (PH) is a pathophysiological disorder that can coexist with numerous clinical entities and can complicate most cardiovascular and respiratory diseases. PH is defined as a mean pulmonary arterial pressure (PAPm) increase ≥ 25mmHg at rest, calculated using right heart catheterization.
There have been a number of significant developments in the PH field in the last 2 years, particularly in available treatments. These advances include the approval of new drugs, new tests for the use of initial drug combination therapies in pulmonary arterial hypertension (PAH), and the approval of drugs for use in chronic thromboembolic PH that is not amenable to surgical thromboendarterectomy.
The field continues to evolve and it is hoped that ongoing clinical trials will identify drugs that can effectively treat PH due to left heart disease and chronic hypoxia. This article highlights the most significant recent advances in PH management.
PAH (group 1) encompasses idiopathic PAH, heritable PAH, and PAH associated with disease, such as connective tissue diseases, HIV infection, portal hypertension, congenital heart disease, schistosomiasis, and that induced by drugs or toxins.1 Despite the considerable effort expended into the research and development of therapeutic agents in the last 20 years, the disease is largely incurable and the general prognosis remains poor. The median survival for an untreated patient is 2.8 years. In the last 3 decades, spectacular advances have been made in the understanding of the molecular mechanisms and signaling pathways involved in the disease, which have resulted in the development of new treatment strategies.
Regarding the new drugs acting on the nitric oxide pathway, whereas phosphodiesterase type 5 inhibitors (PDE-5is) such as sildenafil and tadalafil activate the nitric oxide-cyclic guanosine monophosphate (cGMP) pathway to inhibit cGMP breakdown, its production is promoted by soluble guanylate cyclase stimulators. Treatment of 443 PAH patients (44% and 6% receiving baseline treatment with endothelin receptor antagonists [ERAs] and prostanoids, respectively) with up to 2.5mg riociguat 3 times a day obtained positive results in terms of exercise capacity, hemodynamic parameters, World Health Organization functional class (WHO FC), and time to clinical worsening.2 Exercise capacity increased in both the riociguat and placebo groups. These beneficial effects were maintained for at least 2 years of open follow-up.
The dual ERA antagonist macitentan was evaluated in an event-driven clinical trial that randomized 742 patients to treatment with 3 or 10mg macitentan or placebo during a mean treatment period of 100 weeks.3 The primary endpoint was the time from the initiation of treatment to the first occurrence of a composite endpoint of death, atrial septostomy, lung transplantation, initiation of treatment with intravenous or subcutaneous prostanoids, or worsening of PAH. Macitentan significantly reduced the composite endpoint of morbidity and mortality of patients with PAH and increased exercise capacity. Benefits were seen both in patients not previously receiving therapy for PAH and those receiving additional PAH therapy.
Regarding drugs targeting the prostaglandin pathway, selexipag is an oral selective IP prostacyclin receptor agonist. Although selexipag and its metabolite have similar mechanisms of action to endogenous prostacyclin (IP receptor agonists), they are chemically distinct and have different pharmacological characteristics. A controlled, randomized, event-driven, phase III study including 1156 patients4 showed that selexipag, alone or added to single or dual treatment with ERA or PDE-5i, obtained a 40% reduction (hazard ratio, 0.60; P<.001) in the composite endpoint of morbidity and mortality (which included death from any cause, hospitalization due to worsening of PAH, worsening of PAH that resulted in lung transplantation or atrial septostomy, initiation of parenteral prostanoid therapy or oxygen therapy due to worsening of PAH, and disease progression).
Combination therapy is defined as the simultaneous use of 2 or more classes of drugs. A recent multicenter study, with blinding and a placebo control group, compared initial monotherapy with tadalafil or ambrisentan to initial combination therapy with tadalafil and ambrisentan in patients with de novo PAH and WHO FC II-III.5 The primary composite endpoint was the first event of clinical failure (death, hospitalization, PAH progression, and unsatisfactory clinical response). Positive results were obtained, with a 50% reduction in events in the combination therapy group. In addition, improvements were seen in exercise capacity, rates of a satisfactory clinical response, and the plasma concentration of N-terminal pro–B-type natriuretic peptide.
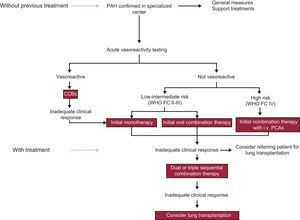
The figure shows a treatment algorithm for patients with PAH that follows the directives of the latest PH guidelines of the European Respiratory Society/European Society of Cardiology.1
. CCBs, calcium channel blockers; i.v., intravenous; PCAs, prostacyclin analogues; WHO FC, World Health Organization functional class.")
The first treatment objective for PH caused by left heart disease (group 2 PH) is to improve the overall treatment of the underlying entity before considering specific measures for PH treatment. Two ongoing multicenter studies are evaluating the treatment of PH caused by left heart disease, the SilHF trial (NCT01616381) with sildenafil and the Melody-1 trial (NCT02070991) with macitentan; the latter study is the only one to require evaluation using right heart catheterization.
Nonetheless, there is no new evidence permitting recommendation of the use of specific treatments for PAH for PH caused by left heart disease, and this is partly due to the absence of studies specifically stratifying patients with PH or specifically targeting this entity.
There is currently no specific treatment for PH associated with chronic pulmonary diseases or hypoxia (group 3 PH). Long-term oxygen therapy partially reduces PH progression in chronic obstructive pulmonary disease. Few data have been published on the specific treatment of PAH and there is still no evidence from controlled studies of drugs for PAH showing symptom or outcome improvements in patients with pulmonary disease.
Pulmonary endarterectomy (PEA) is the treatment of choice for chronic thromboembolic PH (CTEPH; group 4 PH). In Europe, in-hospital mortality is just 4.7%, and it is even lower in high-volume centers. Most patients achieve substantial symptom relief and almost complete normalization of hemodynamic parameters. Riociguat, a soluble guanylate cyclase stimulator, administered for 16 weeks to 261 of 446 patients with inoperable CTEPH or persistent/recurrent PH after pulmonary endarterectomy, achieved a mean increase of 39 meters in the 6-minute walk distance (P<.001, primary endpoint) and a decrease of 246 dyn·s·cm−5 in pulmonary vascular resistance (P<.001, secondary endpoint); the time to clinical worsening was unchanged.6
We conclude by mentioning balloon dilatation of the pulmonary arteries of patients with inoperable CTEPH; this evolving procedure is being performed more and more frequently. Repeat procedures in the same patient decrease pulmonary vascular resistance. Despite the significant reduction in PAPm and improved right ventricular systolic function, the procedure is still being fine-tuned and is limited to specialized centers.