
Urokinase-type plasminogen activator, which is encoded by the PLAU gene, plays a prominent role during collateral arterial growth. We investigated whether the PLAU P141L (C > T) polymorphism, which causes a mutation in the kringle domain of the protein, is associated with coronary collateral circulation in a cohort of 676 patients with coronary artery disease.
MethodsThe polymorphism was genotyped in blood samples using a TaqMan-based genotyping assay, and collateral circulation was assessed by the Rentrop method. Multivariate logistic regression models adjusted by clinically relevant variables to estimate odds ratios were used to examine associations of PLAU P141L allelic variants and genotypes with collateral circulation.
ResultsPatients with poor collateral circulation (Rentrop 0-1; n = 547) showed a higher frequency of the TT genotype than those with good collateral circulation (Rentrop 2-3; n = 129; P = .020). The T allele variant was also more common in patients with poor collateral circulation (P = .006). The odds ratio of having poorly developed collaterals in patients bearing the T allele (adjusted for clinically relevant variables) was statistically significant under the dominant model (odds ratio = 1.83 [95% confidence interval, 1.16-2.90]; P = .010) and the additive model (odds ratio = 1.73 [95% confidence interval, 1.14-2.62]; P = .009).
ConclusionsAn association was found between coronary collateral circulation and the PLAU P141L polymorphism. Patients with the 141L variant are at greater risk of developing poor coronary collateral circulation.
Keywords
Coronary artery disease (CAD) is the leading cause of death in industrialized countries.1–4. Collateral coronary artery growth (coronary arteriogenesis) is a recognized alternative source of blood supply to a myocardial area affected by ischemia in CAD.1,3 Patients with high collateralization have a 36% lower risk of death than those with low collateralization.5 Chronic ischemia and the extent of atherosclerotic burden are important triggers in the etiology of collateral artery formation. Nonetheless, the ability to induce coronary collateral growth is widely heterogeneous in CAD patients, even in those with completely occluded arteries,6 and it may well be a genetically determined process.7,8
Fulton9 demonstrated that the human heart contains an extensive collateral network that is present even before the appearance of obstructive atherosclerotic disease (reviewed by Van Royen et al10). In arteriogenesis, these collateral arterioles interconnect lateral branches proximal and distal to the occluded artery (anastomosis).10,11 Following arterial occlusion, the blood flow is redirected through the preexisting arteriole bridges, which triggers an increase in mechanical forces on the arterial wall, such as shear stress and circumferential stress, resulting from the pressure gradient formed between the high-pressure region proximal to the occlusion, and the low-pressure region located distally.11–13 These forces induce collateral coronary artery growth. This process implies complete structural remodeling of the arterial wall, which involves proliferation of endothelial and smooth muscle cells and ultimately, reorganization of the extracellular component and the internal elastic lamina. Completion of these phases reduces artery wall shear stress to baseline physiologic values.14
Endothelial cells perceive shear stress and transform the signal into changes in gene expression.11–15 The endothelium becomes activated and promotes attraction and adhesion of monocytes, which secrete growth factors and cytokines to the growing arteries.11 In the early stages, collateral arterial growth is associated with an increase in fibroblast growth factor receptor-1 expression at the site of vessel remodeling, while monocytes supply ligands (fibroblast growth factors) via paracrine signaling to promote growth.15 Fibroblast growth factor receptor-1 activation by fibroblast growth factors leads to activation of the Ras/Raf, MEK1/2, ERK1/2 pathway and finally, increased expression of early growth response 1 factor, which is involved in inducing an invasive, migratory phenotype in endothelial and smooth muscle cells. Early growth response 1 factor induces expression of urokinase-type plasminogen activator (u-PA).14
The u-PA is crucial for regulation of cell adhesion, migration, and proliferation,16 for remodeling the artery wall affected by mechanical injury, and for arteriogenesis.16–19 The uPA converts plasminogen into plasmin, which, in turn, activates growth factors, latent forms of cytokines, and matrix metalloproteinases 2 and 9. These are involved in a strictly localized degradation of the extracellular matrix, a critical step to enable smooth muscle cell migration and neointima formation16,17 during collateral vessel growth.
PLAU, the gene that codes for u-PA, is located on chromosome 10q22.2, between 2 regions that have shown a link with Alzheimer disease.20 The single nucleotide polymorphism (SNP) rs2227564, a C/T polymorphism of the second base of codon 141 of the PLAU gene, causes a missense mutation (from proline to leucine) in the kringle domain of u-PA, which has a functional effect on the binding of u-PA to the zymogen of fibrin.21. Although this SNP has been linked to several diseases,22–29 there have been no reports of its association with coronary arteriogenesis. The aim of this study was to evaluate the association between the PLAU P141L polymorphism and collateral coronary artery growth, focusing on the hypothesis that the PLAU L141 variant is associated with a decreased arteriogenic response in patients with CAD.
METHODSStudy Design and Patient SelectionThis study was performed in accordance with the Declaration of Helsinki, and the protocol was approved by the Bioethics Committee of Centre Cardiovascular Sant Jordi and Hospital Universitari de la Vall d’Hebron for the period of 2008 to 2012. All patients gave informed consent and written authorization for participation.
Patients undergoing diagnostic coronary artery catheterization were consecutively selected and enrolled in the study. The cohort comprised of 677 patients with CAD and severe (≥ 70%) stenosis. Patients were excluded if they had a recent (< 1 month) acute myocardial infarction, anemia, recent angioplasty, a previous percutaneous coronary revascularization procedure, coronary artery bypass surgery, infection, inflammation, or chronic renal failure. Each patient's clinical history was studied in detail and the demographic, clinical, and laboratory data were recorded. Hypertension, diabetes mellitus (DM), DM type, hypercholesterolemia, hypertriglyceridemia, smoking history, family history of heart disease, history of angina, type of angina, and acute myocardial infarction were recorded as categorical variables (presence or absence).
Coronary Angiography and Evaluation of the Coronary Collateral CirculationCoronary angiography was performed in multiple orthogonal projections using the Judkins technique. Angiographic evaluation of coronary collateral circulation (CCC) was carried out according to the method of Rentrop et al,30 with the following scale to grade collateral artery filling: 0, no visible filling of collaterals; 1, collateral filling of the side branches of the vessel to be dilated, with no contrast reaching the epicardial segment of the vessel; 2, partial filling of the epicardial segment by collateral vessels, and 3, complete filling of the epicardial segment by collateral vessels.
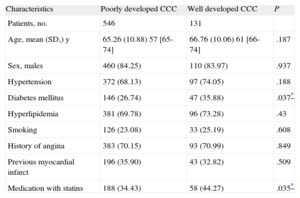
CAD patients were classified according to the grade of CCC as having poorly developed (Rentrop 0-1; n = 546) or well developed (Rentrop 2-3; n = 131) collateral vessels. CCC grade was assessed by 3 experienced interventional cardiologists who were blinded to the patient data. The degree of agreement in the diagnostic evaluation of CCC was high among the 3 observers, as determined by the kappa statistic: κ=0.987; 95% confidence interval (95%CI), 0.953-1.000 (P < .001). Kappa agreement was determined using the first 100 angiograms reviewed.
Genotypic AnalysisBlood extraction was carried out at the time of coronary angiography. Genomic DNA was isolated using the Chemagen automated workstation (Baesweiler, Germany), following the manufacturer's protocol. To determine rs2227564 polymorphism genotypes in blood samples, the TaqMan SNP genotyping assay (Applied Biosystems; Foster City, California, United States) was performed, using the Fast 7900HT (Applied Biosystems) real-time polymerase chain reaction system. Genotyping was repeated in 3 separate determinations for each patient.
Statistical AnalysisContinuous data with a nonnormal distribution were analyzed with the Mann-Whitney U-test. In this study, age was not normally distributed (Shapiro-Wilk, P < .001). Associations between categorical data were assessed with the Fisher exact test or chi-square test, and Hardy-Weinberg equilibrium was calculated with the chi-square test. To estimate the odds ratio (OR) and 95%CI between genotypes and the risk of poor CCC, multivariate logistic regression models were fitted using clinically relevant variables. The interaction terms between the SNP and significant covariates were also analyzed with multivariate regression. Statistical analyses were carried out with the STATA 11.2 software. The same statistical package was used to estimate the power to detect a genetic association.
RESULTSThe statistical analysis showed no significant differences between the 2 groups (poorly developed and well developed CCC) with regard to age, sex, hypertension, hyperlipidemia, smoking, angina, or previous myocardial infarction. The prevalence of DM (35.88%) and the percentage of patients prescribed statins (44.27%) were significantly higher in the poor CCC group (Table 1). In our population, 462 patients (68.24%) had genotype CC, 194 (28.66%) were heterozygous (CT), and 21 (3.01%) had genotype TT. The frequencies of the C and T alleles in the overall study sample were 82.57% and 17.43%, respectively (Table 2). These percentages were consistent with Hardy-Weinberg equilibrium (chi-square, 0.059; P = .8077).
Clinical Characteristics of Patients with Coronary Artery Disease According to the Status of the Coronary Collateral Circulation
| Characteristics | Poorly developed CCC | Well developed CCC | P |
| Patients, no. | 546 | 131 | |
| Age, mean (SD,) y | 65.26 (10.88) 57 [65-74] | 66.76 (10.06) 61 [66-74] | .187 |
| Sex, males | 460 (84.25) | 110 (83.97) | .937 |
| Hypertension | 372 (68.13) | 97 (74.05) | .188 |
| Diabetes mellitus | 146 (26.74) | 47 (35.88) | .037* |
| Hyperlipidemia | 381 (69.78) | 96 (73.28) | .43 |
| Smoking | 126 (23.08) | 33 (25.19) | .608 |
| History of angina | 383 (70.15) | 93 (70.99) | .849 |
| Previous myocardial infarct | 196 (35.90) | 43 (32.82) | .509 |
| Medication with statins | 188 (34.43) | 58 (44.27) | .035* |
CCC, coronary collateral circulation; SD: standard deviation.
Data are expressed as No. (%), mean (standard deviation) or median [interquartile range].
Association of the PLAU P141L Polymorphism (rs2227564) Allelic Variant and Genotype Distribution With Coronary Collateral Circulation in Patients With Coronary Artery Disease
| Poorly developed CCC (n = 546) | Well developed CCC (n = 131) | P | |
| Genotype CC | 359 | 103 | .0160a |
| Genotype CT | 168 | 26 | |
| Genotype TT | 19 | 2 | |
| C allele | 886 | 232 | chi-square 8.0706;P =.004b |
| T allele | 206 | 30 |
CCC, coronary collateral circulation.
P values <.05 were considered statistically significant.
Analysis of PLAU P141L (C > T) polymorphism showed an association between genotype distribution and CCC status (Fisher exact test, P = .0160). The frequency of genotype TT in CAD patients with poorly developed CCC was higher than in those with well developed CCC. The presence of the T allele was also more common in patients with poor CCC (chi-square, 8.0706; P = .004) (Table 2). This association persisted in the subgroup of patients with high-grade stenosis (≥ 95%) (P = .016).
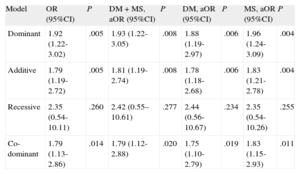
The ORs of having poorly developed CCC in patients carrying the T allele, which codes for the L141 form, were statistically significant in both the dominant genetic model of inheritance (CT+TT vs CC) (OR = 1.92; 95%CI, 1.22-3.02; P = .005) and in the additive model (OR = 1.79; 95%CI, 1.19-2.72; P = .005) (Table 3). After adjustment for potential confounding factors such as DM and medication with statins (Table 1), the ORs of having poor CCC in patients carrying the T allele in the dominant model (CT+TT vs CC) (OR = 1.93; 95%CI, 1.22-3.05; P = .008) and the additive model (OR = 1.81; 95%CI, 1.19-2.74; P = .008) were also statistically significant (Table 3). Similar results were obtained when the data were adjusted separately for DM or for medication with statins (Table 3). Since the difference in deviation between the crude OR and the OR adjusted for DM and/or medication with statins was below 10%, we conclude that these variables cannot be considered confounding factors.
PLAU P141L Single Nucleotide Polymorphism (rs2227564) Odds Ratios for Coronary Collateral Circulation According to Different Models of Inheritance in Patients With Coronary Artery Disease
| Model | OR (95%CI) | P | DM + MS, aOR (95%CI) | P | DM, aOR (95%CI) | P | MS, aOR (95%CI) | P |
| Dominant | 1.92 (1.22-3.02) | .005 | 1.93 (1.22-3.05) | .008 | 1.88 (1.19-2.97) | .006 | 1.96 (1.24-3.09) | .004 |
| Additive | 1.79 (1.19-2.72) | .005 | 1.81 (1.19-2.74) | .008 | 1.78 (1.18-2.68) | .006 | 1.83 (1.21-2.78) | .004 |
| Recessive | 2.35 (0.54-10.11) | .260 | 2.42 (0.55–10.61) | .277 | 2.44 (0.56-10.67) | .234 | 2.35 (0.54-10.26) | .255 |
| Co-dominant | 1.79 (1.13-2.86) | .014 | 1.79 (1.12-2.88) | .020 | 1.75 (1.10-2.79) | .019 | 1.83 (1.15-2.93) | .011 |
95%CI, 95% confidence interval; aOR, adjusted odds ratio; DM, diabetes mellitus; MS, medication with statins; OR, odds ratio.
P values <.05 were considered statistically significant.
Analysis of interactions between the SNP and significant covariates showed no associations. Assuming rates of 18.86% and 11.45% for the presence of the T allele in patients with poorly developed and well developed CCC, respectively, the statistical power to detect genetic differences was 81.6%.
DISCUSSIONAn understanding of the genetic program leading to collateral artery development is important to improve the diagnosis, treatment, and prevention of CAD.7,8 The coronary collaterals can be a useful prognostic marker: patients with scant collateralization have a higher risk of death and should be closely followed. A recent meta-analysis of 12 studies involving 6529 patients showed that CCC is associated with a substantial improvement in survival.5 This study demonstrated that patients with high collateralization have a significantly lower mortality risk than those with low collateralization (relative risk, 0.64; 95%CI, 0.45-0.91; P = .012).
Urokinase induces a local proteolytic cascade that is important for vascular remodeling and angiogenesis, and it is a key participant in the response to cardiovascular injury and in collateral arterial growth. These capabilities make it a promising therapeutic target in vascular diseases.16,18,31 In a flow-induced experimental model of vascular remodeling, u-PA content correlated with neointima growth.32 Similar findings in this line have been reported in arterial tissue studies in transgenic mice17 and in primates.33 In a murine model of hindlimb ischemia, u-PA-deficient mice had a weaker endogenous capacity for restoring blood flow than control mice.19 Traktuev et al34 have demonstrated that u-PA overexpression stimulates vessel growth and tissue perfusion, both of which limit myocardial damage and promote remodeling following an infarction.
On the cell surface, u-PA, a multidomain, multifunctional protein, binds to the high-affinity urokinase receptor (u-PAR)35 located at the leading edge of cell migration, thus enabling regulated proteolysis of extracellular matrix proteins in the direction in which cells are moving. The u-PA/u-PAR complex also regulates vascular remodeling by pathways independent of proteolysis and plasmin generation; for example, by interacting with matrix proteins, integrins, and endocytosis receptors (among others), and by activating intracellular signaling and gene expression to regulate cell adhesion, migration, differentiation, and proliferation.16 Arteriogenesis is also promoted by uPA-mediated leukocyte infiltration, which is not dependent on u-PAR.19
With regard to the PLAU P141L SNP, the change of proline to leucine at amino acid position 121 has been shown to increase the water-repellant ability of this region.21 The proline to leucine substitution may also affect the tertiary structure of the molecule, since P121 is a component of 1 of the 3 antiparallel beta-sheet structures in the kringle domain of u-PA.21 This amino acid change could be a direct or indirect cause of a decreased apparent affinity for fibrin clots.
The ability of u-PA to induce chemotaxis in smooth muscle cells has been attributed to binding of the u-PA kringle domain to the cell surface and the association of u-PA with u-PAR.36 The u-PA can simultaneously bind to 2 cell surface receptors: to u-PAR through the growth factor-like domain and to the Mac-1 integrin through the kringle and proteolytic domains.37 This multicontact, trimolecular complex may play a major role in controlling inflammatory cell migration and in vascular homeostasis.37 Furthermore, the kringle domain of u-PA binds to a specific cell surface receptor that differs from u-PAR36 and contains a sequence that interacts with plasminogen activator inhibitor type 1.38 The kringle domain is implicated in intracellular signaling, induction of cell migration, and cell adhesion.39
In addition to the association between the T variant of u-PA and poorly developed CCC reported here, various studies have shown that the PLAU P141L SNP is involved in certain pathologic conditions, such as neurological disorders and cancer. Of special interest is the role of u-PA in Alzheimer disease.20,23,25,26,40,41 This association is attributed to the functional implication of u-PA in the generation of plasmin, a protease that can degrade beta amyloid proteins. Of note, Riemenschneider et al23 found higher plaque counts in the temporal cortices of patients carrying the PLAU P141L T allele than in those without, and a recent meta-analysis confirmed that presence of the T allele increases the risk of developing Alzheimer disease.26 In other diseases, such as cancer, an association has been observed between genotype CC of PLAU P141L and colorectal cancer invasiveness and metastasis,22 and the SNP has been associated with susceptibility to squamous cell carcinoma of the tongue.27 Furthermore, Begin et al24 observed a correlation between this SNP and u-PA function through the extracellular matrix in the pathogenesis of asthma. Finally, recent reports have shown an association of the P141L SNP with serum lipid concentrations28 and with inflammatory intestinal disease.29
These observations highlight the biological importance of this functional u-PA SNP, which affects the kringle structure of the protein, and are consistent with the association between the T allele in patients with CAD and the poorer CCC detected in this subpopulation. Our results support an implication of the PLAU P141L SNP in the process of arteriogenic growth of the collateral coronary arterioles. It is possible that the L141 variant, which has been shown to affect the structure of the kringle domain of the protein with a subsequent reduction in its affinity for fibrin, may affect the efficiency of extracellular matrix remodeling and the migration capacity of smooth muscle cells. These factors would decrease the arteriogenic response of CAD patients with the T allele variant.
CONCLUSIONSThe findings presented here are the first to show a strong association between the PLAU P141L SNP and CCC. CAD patients with the functional L141 variant show a higher probability of having poorly developed CCC. Although these results should be replicated in additional cohorts, PLAU P141L polymorphism could be a genetic predictor of the arteriogenic response in patients with CAD. Genetic analysis of large samples of cases and controls by genotyping thousands of SNPs distributed over the genome in genome-wide association studies will enable identification of other loci associated with the development of CCC, as has been shown for coronary disease and myocardial infarction (reviewed by Companioni et al42 in 2011). Furthermore, given the central role of u-PA in neointima formation during arterial remodeling, in vitro migration assays using smooth muscle cells from patients with the T allele are needed to determine the functional relevance of this polymorphism. In addition, as suggested by some authors,16 these results support the notion that u-PA gene therapy or improvements in local u-PA production could be promising tools for local stimulation of collateral arterial growth and treatment of myocardial ischemia.
FUNDINGThis study was supported by the Universitat de Barcelona (project ACESBELL 08) and Fundació La Marató de TV3 07 (project 080810).
CONFLICTS OF INTERESTNone declared.
We thank Àlex Cordero, Montse Cairó, Eva Sánchez, Dolors Colell, Teresa Torrent, María José Fernández de Muniain, and Miquel Rugat for their valuable collaboration and technical assistance.