

Risk stratification in pulmonary arterial hypertension (PAH) is essential to provide more aggressive treatment for patients at higher risk. Nevertheless, recently introduced simplified prognostic tools neglect the genetic background. Additionally, pulmonary veno-oclusive disease (PVOD) has never been considered in risk assessment strategies.
MethodsWe analyzed consecutive patients in the Spanish registry of PAH (REHAP) genetically tested, between 2011 and 2022. We applied the 4-strata COMPERA 2.0 model, comparing these results with an amplified score including genetics. Cox regression models were compared using Harrel c-statistics. The application of the model was specifically tested in PVOD before inclusion.
ResultsWe identified 298 patients tested genetically among the group of idiopathic, familial, drug-induced PAH and PVOD patients in the REHAP registry. When we analyzed only patients with all available variables of interest at baseline (World Health Organization functional class, 6-minute walk test, B-type natriuretic peptide or N-terminal pro-B-type natriuretic peptide) and included in the 4-strata model (n=142), after a median follow-up of 58.2 months, 17.6% of patients died and 11.3% underwent lung transplant. The application of the 4-strata model in our population demonstrated a good prognostic capacity (Harrel c of 0.689), which was not improved by the introduction of genetics (c-index 0.690). This last model showed a tendency for a better identification of patients at intermediate-low and intermediate-high risk, and no differences between intermediate-high and high-risk strata.
ConclusionsIn this work, the addition of genetics to the COMPERA 4-strata model achieved a similar global prognostic capacity but changed the identification of different risk strata in a cohort of young genetically tested patients.
Keywords
Pulmonary arterial hypertension (PAH) is a severe and rare disease. Independently of the cause, the sum of endothelial dysfunction, inflammation, vasoconstriction, and cell overgrowth ultimately cause an increase in pulmonary pressures and subsequently right ventricular failure.1 Investigations in this field over the last 2 decades have related alterations in certain molecular pathways involving oxidative stress, inflammation or cell signaling with the development of this disease, among others.2 Variants in the gene encoding bone morphogenetic protein receptor type II (BMPR2) have been found in up to 86% of familial cases and between 14% and 35% of sporadic cases. Alterations in other genes have also been associated with PAH, involving several other distinct functions within the pulmonary endothelium.3 Additionally, pulmonary veno-oclusive disease (PVOD) is an especially infrequent and extremely aggressive form of PAH. Its heritable form is caused by homozygous variants in the gene encoding eukaryotic translation initiation factor 2 alpha-kinase 4 (EIF2AK4),4 and is usually underrecognized and catalogued as an idiopathic form.5 Both the presence of genetic variants in BMPR2 and PVOD have been recognized as individual markers of disease severity.6,7
Despite substantial improvements in survival after the introduction of specific pulmonary vasodilators, the prognosis in PAH is still remarkably poor. Individual risk assessment is essential to provide more aggressive treatment for patients at higher risk of mortality.8 Multiple simple prognostic scores have been designed in the last few years, with comparable results between them.9,10 Nevertheless, none of these scores include the genetic basis or PVOD patients. In this article, we suggest not only including the molecular basis for the correct classification of patients with PAH, but also the need to assess molecular screening in patients with PAH/PVOD, which could further improve the identification of patients at higher risk by adding the value of genetics.
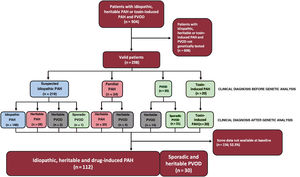
METHODSStudy populationThe Registro Español de Hipertensión Arterial Pulmonar (REHAP) is a prospective registry initiated in January 2007.11 For inclusion in the registry, the diagnosis of PAH required a right heart catheterization with mean pulmonary artery pressure ≥ 25mmHg, pulmonary vascular resistance ≥ 3 WU, and pulmonary artery wedge pressure ≤ 15mmHg. In this study, we included patients older than 18 years with idiopathic, heritable, and toxin-induced PAH, as well as sporadic and heritable PVOD. Genetic testing is routinely offered to patients with idiopathic, familial, drug-associated PAH and PVOD. Only patients with a definitive genetic test result were selected. The study period started in 2011 and ended in February 2021.
PVOD was confirmed if there was a homozygous variant in the gene EIF2AK4, or after histological confirmation. This disease was also diagnosed if there was respiratory impairment after pulmonary vasodilator treatment initiation. Likewise, the condition was considered probable if there was a low diffusing capacity of the lung for carbon monoxide (DLCO) and when at least 2 radiological signs of PVOD out of 3 possible signs were present.12
Written informed consent was obtained in all cases. REHAP and genetic studies were complied in accordance with the principles of the Declaration of Helsinki. The protocol was approved by institutional review boards and ethics committees in all participating centers. Clinical, demographic, analytical, functional, and hemodynamic variables were collected from the REHAP registry.
Genetic analysesGenetic screening of PAH patients started in 2011 through Sanger sequencing and multiplex ligation-dependent probe amplification in BMPR2, TBX4, and KCNK3. This analysis was extended from 2014 to 2020 through a next generation sequencing (NGS) panel of 21 genes (HAP v1.2), which was expanded to cover 35 genes (HAPv3) based on previous research data. The panel was designed with NimbleDesign (Roche, USA). Fragmentation and library preparation were performed with SeqCap EZ Choice Enrichment Kit (Roche, USA) and sequencing was performed with the Illumina MiSeq and NextSeq500 platforms (Illumina, USA). Variant prioritization is detailed in and a custom script developed in-house was applied to analyze copy number variants.13
Later, in 2020, we proceeded to whole-exome sequencing technology. Library preparation was carried out by Agilent SureSelect TM (v 6.0) and all exon kits were followed by sequencing in NovaSeq Sequencer (Illumina, USA). Variant prioritization was also performed by VarSeq (Golden Helix, USA) (figure 1). After variant prioritization, candidate variants were classified according to the American College of Medical Genetics guidelines.14

Genetic counselling was provided for each included patient and first-degree family members. Although a family history was obtained during the first genetic study, a DNA sample for analysis was obtained only from probands. When a genetic variant was detected, the study of first-degree relatives was offered. As a result, segregation analyses were performed in those cases, reclassifying the variable according to the result of this analyses and the presence of other factors of pathogenicity, according to the American College of Medical Genetics guidelines. Pulmonary hypertension was searched for in first-degree relatives with a positive genetic result, by means of transthoracic echocardiography, electrocardiogram, and physical examination. In the first-degree relatives of patients with PVOD, diffusing capacity of the lung for carbon monoxide was also performed.
OutcomesThe main outcome was the first occurrence of lung transplant or death. A Cox regression model was constructed including variables of the validated 4-strata model proposed by the COMPERA registry and validated by the French pulmonary hypertension registry (FPHR).9,15 The original 4-strata model was compared with a model including the same 3 original variables, as well as genetic testing. This last variable was considered positive if there was at least 1 pathogenic (P) or likely pathogenic (LP) variant. Time zero was the date of the first right heart catheterization. The usefulness of the model in PVOD was assessed specifically in this cohort before its inclusion in the global study population. Cut-off points for the 4-strata models were the same, as defined previously.9 To introduce genetics to the COMPERA 2.0 model, we compared the coefficient obtained for this variant in the univariate Cox regression analysis with the mean value of the coefficients obtained for each other variable in the original 4-strata model, similarly to the method used in the original REVEAL study.16 As a result, we considered that the absence of a genetic variable counted as 1 point, and its presence counted as 3 points in the extended model including genetics ().
Statistical analysisCategorical variables are reported as absolute and relative frequencies, and were compared with the Pearson or Fisher exact tests, as appropriate. Continuous variables are reported as means±standard deviation or medians [interquartile range] and were compared with t-tests or Wilcoxon tests, as appropriate. Kaplan-Meier survival curves were compared using the log-rank test. Cox regression models were compared by the results of the Harrel c-statistic using the Somers D test, including only patients with all variables of interest (World Health Organization functional class, 6-minute walk test, B-type natriuretic peptide or N-terminal pro-B-type natriuretic peptide) available at baseline. No imputations were made for missing data. Stata version 14.0 (StataCorp, College Station, USA) and R studio (v 4.0.3) were used for data analysis.
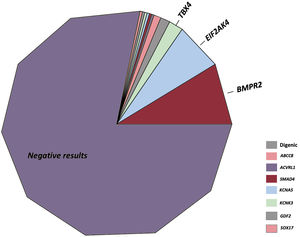
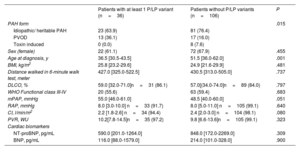
RESULTSResults of genetic testing in the REHAP registryBetween January 2011 and February 2021, we identified 298 genetically tested patients among the group of idiopathic, familial, drug-induced PAH and PVOD patients (figure 1). This group represented 33.0% of the number of individuals under active follow-up in the REHAP registry during this period (298 of 904 patients in the registry with those inclusion criteria). The baseline characteristics of included patients can be seen in . Of these, up to 49.4% of patients without a positive genetic result did not have an evaluation of cardiac biomarkers at baseline. This variable was not available in 39.5% of the group of patients with a P or LP variant. The distance walked in the 6-minute walk test was available in more than 92% of patients in both groups, and functional class was accessible in all the participants (). After genetic analyses, there were 65 patients with at least one P or LP variant (21.8% of the cohort). A variant of unknown significance as the exclusive gene variant was identified in 22 patients (7.4% of the entire cohort). Among the group of patients with at least one P or LP genetic variant, the distribution of genetic variants was as follows: BMPR2 (26 cases), digenic variants including a P or LP in BMPR2 (4 cases), EIF2AK4 (20 cases), TBX4 (6 cases), ABCC8 (3 cases) and other genes (6 cases). In the group of patients with a variant of unknown significance, the most frequent variants were ABCC8 (5 cases) and NOTCH3 (4 cases). There were also variants in the following genes: BMPR1B, CAV1 (x 2), CBLN2, CPS1, CYP1A1, EIF2AK4, ENG, GDF2, KCNA5 (x 2), KCNK3, and SMAD9 classified as variant of unknown significance ().
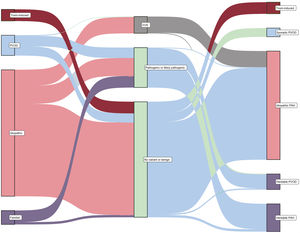
Based on the clinical background, 219 cases were classified initially as idiopathic PAH. After genetic analysis, 31 cases were reclassified as other subtypes of pulmonary hypertension (14.2%), most of them being reclassified as heritable PAH due to the presence of a P or LP variant. Of 24 cases initially classified as familial PAH, the vast majority of cases were confirmed as heritable PAH (83.3%), but 4 cases were reclassified as heritable PVOD due to the discovery of a homozygous EIF2AK4 variant (16.7%). Finally, of the 41 patients with clinically suspected PVOD, genetic testing allowed the reclassification and diagnosis of heritable PVOD in 14 of them (34.1%) (figure 2 and figure 3).


Considering only those patients with available baseline variables and included in the 4-strata model (n=142), the median age was 46.0 years (34.0-59.0). In comparison with patients without significant genetic variants (n=106), patients with a P or LP variant were significantly younger (36.5 vs 51.5 years; P=.001). Additionally, patients with P or LP variants had slightly worse hemodynamic parameters (table 1). PVOD was relatively more prevalent in the group of patients with P or LP genetic variants. In comparison with patients with sporadic PVOD, those with heritable PVOD showed younger age at diagnosis, better functional class, and lower levels of cardiac biomarkers, but hemodynamic severity was similar ().
Baseline characteristics of included patients
| Patients with at least 1 P/LP variant (n=36) | Patients without P/LP variants (n=106) | P | |
|---|---|---|---|
| PAH form | .015 | ||
| Idiopathic/ heritable PAH | 23 (63.9) | 81 (76.4) | |
| PVOD | 13 (36.1) | 17 (16.0) | |
| Toxin induced | 0 (0.0) | 8 (7.6) | |
| Sex (female) | 22 (61.1) | 72 (67.9) | .455 |
| Age at diagnosis, y | 36.5 [30.5-43.5] | 51.5 [36.0-62.0] | .001 |
| BMI, kg/m2 | 25.8 [23.2-29.6] | 24.9 [21.6-29.9] | .481 |
| Distance walked in 6-minute walk test, meter | 427.0 [325.0-522.5] | 430.5 [313.0-505.0] | .737 |
| DLCO, % | 59.0 [32.0-71.0]n=31 (86.1) | 57.0[(34.0-74.0]n=89 (84.0) | .797 |
| WHO Functional class III-IV | 20 (55.6) | 63 (59.4) | .683 |
| mPAP, mmHg | 55.0 [46.0-61.0] | 48.5 [40.0-60.0] | .051 |
| RAP, mmHg | 8.0 [3.0-10.0] n=33 (91.7) | 8.0 [5.0-11.0] n=105 (99.1) | .640 |
| CI, l/min/m2 | 2.2 [1.8-2.6] n=34 (94.4) | 2.4 [2.0-3.0] n=104 (98.1) | .080 |
| PVR, WU | 10.2[7.8-14.5]n=35 (97.2) | 9.8 [6.6-13.6]n=105 (99.1) | .323 |
| Cardiac biomarkers | |||
| NT-proBNP, pg/mL | 590.0 [201.0-1264.0] | 848.0 [172.0-2269.0] | .309 |
| BNP, pg/mL | 116.0 [88.0-1579.0] | 214.0 [101.0-328.0] | .900 |
BMI, body mass index; CI, cardiac index; DLCO, diffusing capacity of the lung for carbon monoxide; LP, likely pathogenic; mPAP, mean pulmonary artery pressure; NT-proBNP, N-terminal pro-B-type natriuretic peptide; P, pathogenic; PAH, pulmonary arterial hypertension; PVOD, pulmonary veno-occlusive disease; PVR, pulmonary vascular resistance; RAP, right atrial pressure; WHO, World Health Organization.
The data are expressed as No. (%) or median [interquartile range].
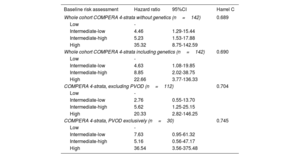
After a median follow-up of 58.2 [31.4-90.7] months, 17.6% of patients died and 11.3% underwent lung transplant. When PVOD was specifically tested, the application of the 4-strata COMPERA model resulted in a similar goodness-of-fit in that cohort in comparison with the rest of the study population (Harrel c of 0.745 in PVOD vs 0.704 in the global cohort excluding PVOD) (table 2). Globally, the application of the 4-strata risk score criteria in our population demonstrated a good prognostic capacity (Harrel c of 0.689). The introduction of the genetic result did not achieve a better prognostic capacity evaluated by the c-statistic (C-index of 0.690 for the extended model adding genetics, P=.367). The use of genetics showed a nonsignificant trend toward the identification of patients at intermediate-low and intermediate-high risk (P=.053) (figure 4). The model including genetics showed no statistical differences between intermediate-high and high-risk patients (P=.114 with the model using genetics vs P <.001 in the original model) (figure 4, and ).
Cox regression models of included patients
| Baseline risk assessment | Hazard ratio | 95%CI | Harrel C |
|---|---|---|---|
| Whole cohort COMPERA 4-strata without genetics (n=142) | 0.689 | ||
| Low | - | ||
| Intermediate-low | 4.46 | 1.29-15.44 | |
| Intermediate-high | 5.23 | 1.53-17.88 | |
| High | 35.32 | 8.75-142.59 | |
| Whole cohort COMPERA 4-strata including genetics (n=142) | 0.690 | ||
| Low | - | ||
| Intermediate-low | 4.63 | 1.08-19.85 | |
| Intermediate-high | 8.85 | 2.02-38.75 | |
| High | 22.66 | 3.77-136.33 | |
| COMPERA 4-strata, excluding PVOD (n=112) | 0.704 | ||
| Low | - | ||
| Intermediate-low | 2.76 | 0.55-13.70 | |
| Intermediate-high | 5.62 | 1.25-25.15 | |
| High | 20.33 | 2.82-146.25 | |
| COMPERA 4-strata, PVOD exclusively (n=30) | 0.745 | ||
| Low | - | ||
| Intermediate-low | 7.63 | 0.95-61.32 | |
| Intermediate-high | 5.16 | 0.56-47.17 | |
| High | 36.54 | 3.56-375.48 | |
95%CI, 95% confidence interval; PVOD, pulmonary veno-oclusive disease.
 and including genetics (B). Log-rank test P <.001 for both models.")
The identification of a pathogenic or LP variant in genes related to the development of PAH or PVOD was associated with multiple reclassifications from one PAH group to another. In a cohort of young patients with PAH or PVOD, the 4-strata COMPERA model did not demonstrate good differentiation of patients at intermediate-low and intermediate-high risk. The inclusion of the genetic background showed a tendency for a better discrimination of patients at intermediate risk (figure 5).
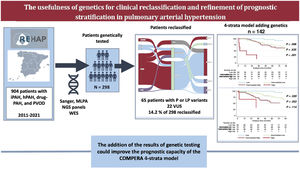
 with idiopathic, heritable, and drug-induced PAH, as well as PVOD during the 2011-2021 period. Two-hundred ninety-eight patients were tested genetically. P or LP variants were found in 65 of those 298 patients, and consequently 14.2% of those were reclassified clinically. The addition of the result of genetic tests did not improve the global prognostic capacity of the 4-strata model based on COMPERA 2.0. hPAH, heritable pulmonary arterial hypertension; iPAH, idiopathic pulmonary arterial hypertension; NGS, next generation sequencing; LP, likely pathogenic variant; P, pathogenic variant; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; PVOD, pulmonary veno-occlusive disease; VUS, variant of unknown significance; WES, Whole-Exome sequencing.")
Central illustration. A summary of the main findings of the study are shown in the figure. On the left panel there is a summary of the total population of the Spanish registry of pulmonary hypertension (REHAP) with idiopathic, heritable, and drug-induced PAH, as well as PVOD during the 2011-2021 period. Two-hundred ninety-eight patients were tested genetically. P or LP variants were found in 65 of those 298 patients, and consequently 14.2% of those were reclassified clinically. The addition of the result of genetic tests did not improve the global prognostic capacity of the 4-strata model based on COMPERA 2.0. hPAH, heritable pulmonary arterial hypertension; iPAH, idiopathic pulmonary arterial hypertension; NGS, next generation sequencing; LP, likely pathogenic variant; P, pathogenic variant; PAH, pulmonary arterial hypertension; PH, pulmonary hypertension; PVOD, pulmonary veno-occlusive disease; VUS, variant of unknown significance; WES, Whole-Exome sequencing.
An early analysis after the introduction of genetic testing for patients in REHAP registry identified several cases associated with genetic variants in the BMPR2, KCNK3 and TBX4 genes, which were associated with a different survival profile.17 Furthermore, the identification of EIF2AK4 as the cause of heritable PVOD was one of the most remarkable findings in the field of genetics in PAH.4 A relatively high prevalence of heritable PVOD was described in the REHAP population, especially in patient of Romani race. The discovery of a highly preserved homozygous variant in this extremely inbred population suggested a possible founder effect.18 The present study represents a continuation of these efforts to associate clinical phenomena and genetics. Nowadays, it is more common to perform genetic testing with highly accurate techniques that have increased the genetic diagnostic yield. More importantly, the number of genes identified in these patients have multiplied, which has allowed us to reclassify several cases according to the genetic results (figure 2, ).
Previous studies demonstrated a significant prevalence of genetic variants in patients with PAH associated with exposure to fenfluramine.19 In this line, some authors have proposed a possible genetic mechanism in patients with PAH associated with previous exposure to methamphetamines.20 Subsequently, current recommendations suggest the use of genetic testing for those patients with PAH related to previous exposure to toxins or drugs. In our study, we did not discover any genetic variants in this group. Therefore, genetics did not influence the clinical classification of these patients. Nevertheless, it is important to note that all patients in this group had previous exposure to toxic rapeseed oil, representing the largest cohort studied so far.
Compared with previous registries studying baseline risk, such as the COMPERA 4-strata,9 the REVEAL Lite 2.010 or the simplified model based on the FPHR,21 the application of the 4-strata model in the REHAP genetically tested population showed a similar goodness-of-fit evaluated by the Harrel c-statistic. This C-index was comparable to those of the other registries. Nonetheless, the identification of patients at intermediate-low and intermediate-high risk was suboptimal using the original model (P=.635). In contrast, there was a tendency for better discrimination of intermediate-low and intermediate-high risk using the extended model including genetics (P=.053). There were no statistical differences between intermediate-high and high-risk strata with this last model (figure 5). Considering the identification of genetic variants in PAH and its possible association with prognosis, a previous meta-analysis including approximately 700 patients demonstrated that the presence of P or LP variants in the gene encoding BMPR2 was associated with prognostic value.6 Hemodynamic severity was thought to play a key role in the higher mortality observed in those patients with variants in BMPR2, especially in younger patients. Although the prognostic impact of other genes has not yet been evaluated, there have been reports of the influence of genetic variants of other genes on hemodynamics.3 In our study, mean pulmonary artery pressure was slightly higher and the cardiac index was slightly lower in those patients with P or LP genetic variants. Moreover, these hemodynamic differences seemed not to be clinically relevant, so other possible mechanisms could be associated with better risk stratification using genetics. A possible explanation of this finding is that the age of included patients in our study was significantly lower compared with that in patients included in the COMPERA 2.0 (65.7 years)9 and those included in the FPHR, a registry that validated the 4-strata model externally (61±15 years).15 Indeed, our population resembles that included in Cluster 1 of the COMPERA registry, a population without comorbidities and characterized by its hemodynamic severity.22 It is possible that in Cluster 1 patients, additional factors might be necessary to better stratify patients at intermediate risk. The genetic fingerprint could have influenced the stratification at intermediate risk since heritable cases usually have longer disease duration and more frequently require lung transplant. Additionally, the introduction of patients with heritable PVOD could have been linked to the relative prognostic capacity of genetics in young patients. Nevertheless, a similar number of patients with sporadic and heritable PVOD were included, and the former were significantly older, had a worse functional class and higher cardiac biomarker levels, aspects that are also associated with worse survival.
Another highly interesting aspect of our study is the inclusion of PVOD. Thirty-five patients were clinically classified as PVOD after initial work-up. Of them, 14 were reclassified as heritable after the identification of a homozygous variant in EIF2AK4 (40.0%). Compared with previous data by Eyries et al.,4 the diagnostic yield of genetic analysis in those patients with sporadic PVOD was slightly superior in our study (25.0% in their study). Thus, it is important to note that in their study all cases were histologically proven. In our series, of 20 patients with heritable PVOD (confirmed genetically), 9 underwent lung transplant (45.0%) and 5 died during follow-up (25.0%). In contrast, 5 of 22 patients in the group with sporadic PVOD underwent lung transplant (22.7%) and 8 died (36.4%). In all transplanted patients and in 5 of 8 nonsurvivors with sporadic PVOD, there was histological confirmation. Although these patients could be associated with a specific phenotype, which includes the development of pulmonary edema after the initiation of pulmonary vasodilators, this rare condition is usually catalogued as idiopathic PAH.23 As shown in our study, the reclassification of patients from an initial PAH group to heritable PVOD is relatively frequent. We included a significant number of patients with sporadic PVOD, who had similar characteristics to those with heritable PVOD, but differed broadly in some other important features. Therefore, those patients with sporadic PVOD were older and had a strikingly higher N-terminal pro-B-type natriuretic peptide level at diagnosis, as previously described by Montani et al.24 (median age 35.0 years in our study vs 26.0 including pediatric patients in the work by Montani et al. in heritable PVOD; 55.0 years vs 60.0 in sporadic PVOD). Bearing this in mind, the inclusion of PVOD seems to be relevant not only to determine whether the presence of a definitive genetic result could be associated with worse outcomes in patients initially classified as idiopathic PAH, but also in those patients with an initial clinical suspicion of PVOD. This is the first time that PVOD patients have been included in a multivariable model validated for patients with PAH. To test the value of those models in PVOD, we performed them specifically in the PVOD population, showing that the 4-strata COMPERA 2.0 model could also be useful for this population. Moreover, specific models should be carried out in patients diagnosed with PVOD in the future to verify our results.
The 4-strata model was recently introduced in the 2022 pulmonary hypertension guidelines of the European Society of Cardiology/European Respiratory Society as the preferred tool for prognostic stratification at follow-up.25 Hypothetically, the addition of genetics, as a noninvasive, static variable usually determined after the initial diagnosis, could allow us to better stratify patients during follow-up. The study was not designed to detect differences between different risk groups in PAH and PVOD patients. Nevertheless, considering the findings in our work, a larger sample might have found differences between risk strata in PAH and PVOD.
LimitationsSome limitations could limit the results of our work. First, because we included patients who had undergone a genetic test exclusively, a selection bias could favor the inclusion of young patients with heritable PAH and PVOD. Second, we included a limited number of patients from the registry due to the presence of several missing values in cardiac biomarkers and the limited number of genetically screened patients. Nevertheless, the inclusion rate was similar to that in the study by COMPERA 2.0 investigators that introduced the 4-strata model (142 patients from 904 possible candidates in our study; 1655 of an initial cohort of 10 825 patients in COMPERA).9 Finally, we intentionally did not include prognostic variables such as right ventricular function or cardiopulmonary exercise tests, since the main aim of the study was to compare the prognostic value of previously validated simplified models with our model including genetic testing at baseline.
CONCLUSIONSThis study provides new hypotheses regarding the usefulness of genetic testing for clinical reclassification and prognosis. Even though the addition of genetic results did not improve the value of the current 4-strata model, this study suggests that the use of genetics might lead to more accurate stratification of intermediate risk not only in the subset of young patients with idiopathic, heritable, drug-induced PAH, but also in patients with heritable or sporadic PVOD.
FUNDINGThis project was funded by the projects “Bases Genético-Moleculares de la Medicina de Precisión en la Hipertensión Arterial Pulmonar” and “Moving toward to a -omic classification for pulmonary arterial hypertension”, Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación, Gobierno de España. Award numbers: PI 18/01233, PI21-01593, and PI21-01690. A. Cruz-Utrilla holds a research-training contract “Rio Hortega” (CM20/00164) from the Spanish Ministry of Science and Innovation (Instituto de Salud Carlos III).
AUTHORS’ CONTRIBUTIONSJ. Antonio Tenorio-Castaño and P. Escribano-Subias contributed equally in this article. Substantial contributions to the conception or design of the work; or the acquisition, analysis, or interpretation of data for the work: A. Cruz-Utrilla, N. Gallego-Zazo, C. Pérez-Olivares, I. Hernández-González, P. Bedate, A. Martínez Meñaca, M. López Meseguer, P. Lapunzina, M. Pérez Núñez, N. Ochoa Parra, J. Tenorio-Castaño, P. Escribano-Subias. Drafting the work or revising it critically for important intellectual content: A. Cruz-Utrilla, N. Gallego-Zazo, J. Tenorio-Castaño, P. Escribano-Subias. Final approval of the version to be published: A. Cruz-Utrilla, N. Gallego-Zazo, C. Pérez-Olivares, I. Hernández-González, P. Bedate, A. Martínez Meñaca, M. López Meseguer, P. Lapunzina, M. Pérez Núñez, N. Ochoa Parra, J. Tenorio-Castaño, P. Escribano-Subias. Agreement to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved: A. Cruz-Utrilla, N. Gallego-Zazo, C. Pérez-Olivares, I. Hernández-González, P. Bedate, A. Martínez Meñaca, M. López Meseguer, P. Lapunzina, M. Pérez Núñez, N. Ochoa Parra, J. Tenorio-Castaño, P. Escribano-Subias.
CONFLICTS OF INTERESTA. Cruz-Utrilla has received funding from MSD, Janssen, Gossamer Bio, and Ferrer. N. Gallego Zazo did not receive funding. C. Pérez-Olivares has received funding from Janssen and MSD. I. Hernández-González did not receive funding. P. Bedate did not receive funding. A. Martínez Meñaca has received funding from MSD, Janssen, Gossamer Bio, AOP Orphan, Chiesi, Rovi, and Leo Pharma. M. López Meseguer has received funding from MSD, Janssen, and Ferrer. P. Lapunzina did not receive funding. M. Pérez Núñez did not receive funding. N. Ochoa Parra did not receive funding. D. Valverde did not receive funding. J. Tenorio-Castaño did not receive funding. Jair Tenorio received a grant from FEDER and FCHP. P. Escribano-Subias has received funding from MSD, Janssen, Gossamer Bio, AOP and Ferrer.
.
This project was supported by the Spanish pulmonary arterial hypertension registry (REHAP), and Centros de Investigación Biomédica en Red de Enfermedades Cardiovascular (CIBERCV), Enfermedades Raras (CIBERER), and Enfermedades Respiratorias (CIBERES), all of them linked to the Instituto de Salud Carlos III (Spanish Ministry of Science and Innovation). In addition, we would like to thank our patients for making this study possible.
Supplementary data associated with this article can be found in the online version, at https://doi.org/10.1016/j.rec.2022.11.002