Palabras clave
INTRODUCCIÓN
La mayoría de los aneurismas ventriculares (AV) izquierdos son adquiridos como resultado de un infarto de miocardio (IM)1; otras posibles causas son la miocardiopatía hipertrófica (MCH) con obstrucción medioventricular2, el traumatismo/cirugía cardíaca, la tuberculosis, la enfermedad de Chagas, el origen anómalo de la arteria coronaria izquierda en la arteria pulmonar, la fiebre reumática, la sarcoidosis y la miocarditis1,3.
El aneurisma congénito de ventrículo izquierdo (VI) es una entidad muy rara que se diagnostica en su mayoría por exclusión, una vez descartadas otras posibles etiologías1. Su presentación clínica es muy variable y su tratamiento no está estandarizado debido a su baja prevalencia, por lo que, en ocasiones, resulta controvertido en particular en lo que se refiere a las indicaciones de tratamiento quirúrgico. Presentamos el caso de una mujer con un AV izquierdo congénito que requirió resección quirúrgica.
CASO CLÍNICO
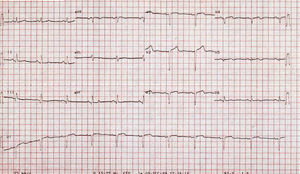
Mujer de 49 años sin historia familiar de cardiopatía, con antecedentes de taquicardias supraventriculares paroxísticas desde los 17 años. A los 34 años fue estudiada por un cuadro de diplopía con sospecha de accidente isquémico transitorio (AIT) sin otra clínica cardiovascular, y se detectó en el electrocardiograma (ECG) una imagen de necrosis inferior y anteroseptal antigua (fig. 1). La radiografía de tórax mostraba una calcificación sobre el ápex cardíaco y una ventriculografía con isótopos radiactivos con 99m Tc objetivó ya entonces una imagen aneurismática en pared inferior.

Fig. 1. Electrocardiograma de 12 derivaciones que muestra una imagen de necrosis inferior y anteroseptal antigua.
A los 44 años, sin presentar sintomatología cardiovascular, fue valorada en el servicio de cardiología por un ECG patológico. Se realizó una ergometría, que mostró una buena capacidad funcional (8 MET), un descenso del segmento ST de 1 mm en la cara inferior con el esfuerzo máximo y extrasistolia ventricular (EV) frecuente. El Holter presentaba extrasistolia supraventricular escasa con fenómenos de repetición, EV muy frecuente con parejas y una racha de taquicardia ventricular no sostenida. El ecocardiograma transtorácico (ETT) inicial no detectó alteraciones. Una tomografía de perfusión con talio objetivó un amplio defecto de perfusión en la cara anterior, apical e inferior, reversible en reposo, salvo en la zona apical. Para aclarar estos hallazgos discordantes se realizó un cateterismo cardíaco, que mostró unas coronarias normales y una masa aneurismática apical ventricular izquierda calcificada.
Desde entonces la paciente fue seguida ambulatoriamente, sin presentar clínica cardiovascular bajo tratamiento médico.
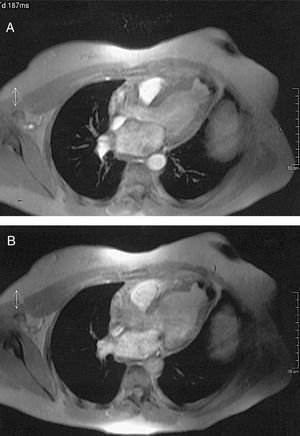
Dado que la mala ventana ultrasónica dificultaba la caracterización del aneurisma, se realizó una resonancia magnética (RM) cardíaca que demostró un aneurisma en el ápex de VI de 257 mm2 de área y un perímetro de 81 mm con calcificación de su pared (fig. 2). En ETT de control posteriores se observó una tendencia a la dilatación de cavidades ventriculares izquierdas (DTDVI 62,3/DTSVI 41,6 mm) con preservación de la función sistólica (fracción de eyección del ventrículo izquierdo del 61%).

Fig. 2. Imagen de cinerresonancia magnética turbo eco de gradiente (TR = 11 ms, TE = 4 ms, a = 20o) cardíaca en la que se objetiva el aneurisma apical ventricular izquierdo con su pared calcificada en sístole (A) y diástole (B).
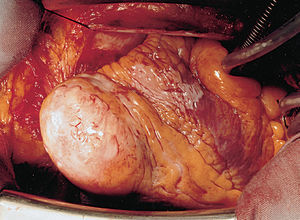
Ante la evidencia de datos indicativos de remodelado ventricular, la paciente fue finalmente intervenida, y se le realizó una aneurismectomía y una endoaneurismorrafia tipo Dör (fig. 3), confirmándose el diagnóstico de AV en el estudio anatomopatológico. La evolución ha sido favorable en el seguimiento a 24 meses, bajo tratamiento con bloqueadores beta, anticoagulación oral e inhibidores de la enzima de conversión de la angiotensina (IECA), con normalización de los diámetros ventriculares en los ETT postoperatorios.

Fig. 3. Pieza de aneurismectomía.
DISCUSIÓN
El AV congénito es una entidad clínica infrecuente (el 0,4% de 750 necropsias cardíacas)4 que se diagnostica una vez descartadas otras causas etiológicas más prevalentes. Es importante distinguirlo del divertículo congénito de VI (DCVI), entidad con la que con frecuencia se confunde5. Éste se caracteriza por presentar un cuello de comunicación con la cavidad ventricular estrecho, una pared compuesta por tres capas diferenciadas y una contracción sistólica sincrónica con el ventrículo. Por el contrario, en los aneurismas, la zona de unión al VI es ancha, histológicamente carecen de capa muscular miocárdica y presentan una única de tejido fibroelástico, en ocasiones calcificada6; asimismo, muestran durante la sístole una expansión paradójica5. Además, mientras los AV se presentan como defectos congénitos aislados, el 70% de los DCVI se asocian con defectos congénitos de la línea media toracoabdominal y malformaciones cardíacas congénitas7.
La severidad de las manifestaciones clínicas de los AV congénitos varía ampliamente de unos pacientes a otros3, incluidas las arritmias supraventriculares y ventriculares (como nuestro caso)8,9, la insuficiencia cardíaca, los embolismos periféricos, la endocarditis, la rotura o el taponamiento cardíaco4, o incluso la muerte súbita3.
La resección quirúrgica está indicada en casi todos los pacientes para prevenir estas complicaciones7,8, aunque algunos grupos defienden una actitud conservadora en los pacientes asintomáticos con medidas destinadas a la prevención de la endocarditis y los embolismos con antiagregación o anticoagulación oral3.
En nuestro caso, la progresiva dilatación de las cavidades ventriculares, junto con los antecedentes de un cuadro neurológico de posible origen embólico, llevó a plantearse la cirugía como la opción terapéutica de elección para prevenir futuras complicaciones, a pesar de tratarse de una paciente prácticamente asintomática.
La presencia de arterias coronarias normales, la exclusión de IM previo o MCH, como confirmó la histología de la pieza resecada, así como de otras posibles causas etiológicas en su historial clínico, nos permitió inferir que el origen más probable del AV era congénito, lo cual se veía apoyado por el inicio de las manifestaciones clínicas en una edad temprana y la presencia de calcificación en la región aneurismática, que requiere cierto tiempo para establecerse.
Varias técnicas de imagen son útiles para el diagnostico de los AV, incluidas la radiografía de tórax, la ecocardiografía simple o de contraste, el cateterismo cardíaco y la RM3,4,7,10. En el presente caso, debido a una mala ventana ecocardiográfica, el diagnóstico del aneurisma pasó inadvertido en las exploraciones ecocardiográficas iniciales y la RM fue la técnica que objetivó con mayor precisión la morfología y la función cardíacas, permitiendo el diagnóstico correcto e inocuo del AV apical calcificado.
Correspondencia: Dr. R. Pérez-Fernández.
Servicio de Cardiología. Complexo Hospitalario de Pontevedra.
Mourente-Montecelo. 36071 Pontevedra. España.
Correo electrónico: rula_perez@hotmail.com