ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2022 - El Congreso de la Salud Cardiovascular

Palma de Mallorca y online,
20 - 22 de Octubre de 2022
Introducción
Dr. Juan José Gómez Doblas
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
6055. Cardiopatías familiares: miscelánea
Fecha
: 22-10-2022 13:45:00
Tipo
: Pósteres
Sala
: E-poster 3 (Planta 0)
6055-8. DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
Yolanda Rico Ramírez1, Laura Torres-Juan1, Francisca Ramis Barceló2, Jaume Pons Llinares1, Elena Fortuny Frau1, Rafael Félix Ramis1, Víctor J. Asensio1, Icíar Martínez1, Vicente Peral Disdier1 y Damián Heine-Suñer1
1Hospital Son Espases, Palma de Mallorca y 2Clínica Planas, Palma de Mallorca.
1Hospital Son Espases, Palma de Mallorca y 2Clínica Planas, Palma de Mallorca.
Introducción y objetivos: El gen NOTCH1 (cromosoma 9q34) codifica la proteína NOTCH1, fundamental en el desarrollo cardiaco cuyas mutaciones se han asociado a defectos congénitos. Las variantes más comúnmente descritas son de un solo nucleótido, con penetrancia incompleta (75%) y expresión variable. Hay pocas deleciones descritas y la patogenicidad de la haploinsuficiencia de NOTCH1 es poco conocida. Describimos el primer caso de deleción exclusiva en NOTCH1 como causa de aneurisma aórtico (DLAo) en paciente con válvula aórtica (VAo) tricúspide.
Métodos: Realizamos estudio genético del caso índice utilizando secuenciación del genoma completo y análisis de variantes con Illumina Trusight Software Suite (TSS). Se realizó revisión bibliográfica de los hallazgos genéticos en los casos descritos previamente.
Resultados: Mujer de 43 años, sospecha de síndrome de Marfan (SM) y disnea de esfuerzo. Presentaba pectus carinatum, aumento de la envergadura de los grupos y escoliosis, sin cumplir criterios diagnósticos de SM (score sistémico < 7 puntos). El ecocardiograma (ETT) reveló DLAo (43 mm) y doble lesión aórtica con estenosis leve e insuficiencia grave con dilatación ventricular izquierda moderada. Fue intervenida con la técnica de Bentall-Bono con éxito. Como hallazgos intraoperatorios destacó VAo trivalva altamente desestructurada y tejido aórtico muy frágil. El estudio histológico mostró necrosis quística de la capa media, hallazgo típico en DLAo degenerativa. Seguimiento a los 2 años sin complicaciones, aunque en tomografía de control se objetivó dilatación de arteria pulmonar (40 mm). El test genético con exoma clínico sugirió una deleción en NOTCH1 que se confirmó con estudio de secuencia del genoma completo y análisis de variantes con TSS. No se encontraron otras variantes potencialmente patógenas relacionadas. La deleción medía 117 kb en 9q34,3 e incluía gran parte del gen NOTCH1 (desde 5'-UTR hasta el exón 18) sin ningún otro gen codificante. Su padre ya fallecido había sido portador de VAo protésica. Su hermana no presenta fenotipo ni es portadora de la mutación.
|
Comparación fenotipo-genotipo entre pacientes previamente descritos con deleción menor de 1Mb que incluye el gen NOTCH1 y nuestro paciente |
|||
|
Paciente |
Fenotipo cardiaco |
Tamaño de la deleción |
Genes incluidos en la deleción |
|
A2 6 |
Válvula aórtica bicúspide |
0,22 Mb |
3 genes OMIM (NOTCH1, INPP5E y PMPCA) y dos genes RefSeq (SEC16A y C9orf163) |
|
A3 6 |
Válvula aórtica bicúspide y coartación de aorta con ventrículo izquierdo de tamaño y función sistólica conservada. |
0,22 Mb |
Tres genes OMIM (NOTCH1, INPP5E y PMPCA) y dos genes RefSeq (SEC16A y C9orf163) |
|
B1 6 |
Corazón izquierdo hipoplásico, válvula mitral displásica, VD de doble salida e hipoplasia tubular del arco aórtico izquierdo |
614,3 kb |
NOTCH1 y tres genes OMIM (AGPAT2, ABCA2 y MAN1B1). La deleción también contiene 22 genes RefSeq (SEC16A, EGFL7, MIR126, LCN10, LCN6, LCN8, C9orf86, PHPT1, EDF1, TRAF2, FBXW5, C8G, LCN12, PTGDS, CLIC3, ABCA2, FUT7, NPDC1, ENTPD2 y C9orf140). |
|
1-II-3 3 |
Síndrome de Adams-Oliver con arterias pulmonares estenóticas |
85 Kb |
Región 5’ de NOTCH1 (promotor y exón 1) |
|
Nuestro paciente |
Doble lesión aórtica sobre válvula aórtica tricúspide con estenosis leve e insuficiencia grave, dilatación ventricular izquierda moderada, dilatación de aorta ascendente (43 mm) |
117 kb |
Gran parte de NOTCH1 (hasta el exón 18), así como tres genes de ARN no codificantes (MIR4673, MIR4674, NALT1 y LINCO1451). |
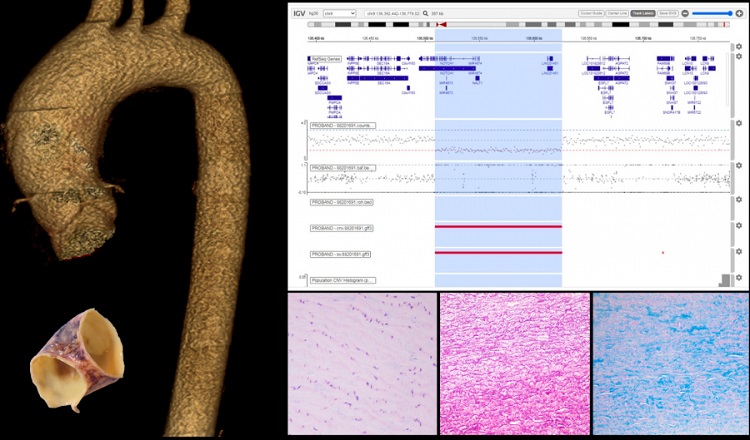
- Reconstrucción 3D de la aorta. B. Deleción. C. Segmento macroscópico de arteria aórtica. D. Hallazgos histológicos: degeneración de la capa media con pérdida de celularidad, fibras elásticas fragmentadas y necrosis quística.
Conclusiones: Se trata de un caso de DLAo no sindrómica y VAo tricúspide, en la que se detecta haploinsuficiencia de NOTCH1 como único gen alterado. Describimos por primera vez una deleción que afecta exclusivamente a NOTCH1 con DLAo torácica y sin VAo bicúspide, apoyando la patogenicidad de la haploinsuficiencia NOTCH1.
Comunicaciones disponibles de "Cardiopatías familiares: miscelánea"
- 6055-1. MODERADORA
Helena Llamas Gómez, Sevilla (Córdoba)
- 6055-2. KCNH2 P. GLY262ALAFSTER98: UNA NUEVA VARIANTE ASOCIADA AL SÍNDROME DE QT LARGO EN UNA COHORTE ESPAÑOLA
- Alejandro Junco Vicente, Alicia Pérez Pérez, Elías Cuesta Llavona, Miguel Soroa Ortuño, Noemi Barja González, Yván Rafael Persia Paulino, José Manuel Rubín López, José Julián Rodríguez Reguero, Bárbara C. Fernández Barrio, Eliecer Coto García, César Morís de la Tassa, Juan Gómez de Oña y Rebeca Lorca Gutiérrez
Hospital Universitario Central de Asturias, Oviedo (Asturias).
- 6055-3. MANIOBRAS ELECTROCARDIOGRÁFICAS EN EL DESPISTAJE DE CANALOPATÍAS: ¿CUÁL ES LA RENTABILIDAD DIAGNÓSTICA?
- María Maeve Soto Pérez1, Jesús Piqueras Flores2, Jorge Martínez del Río1, Pedro Pérez Díaz3, María Aránzazu González Marín4, Martín Negreira Caamaño1, Cristina Mateo Gómez1, Daniel Águila Gordo1, Andrez Felipe Cubides Novoa1, Pablo Soto Martín1, Emilio Blanco López1, José María Arizón Muñoz1 y Javier Jiménez Díaz5
1Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 2Unidad de Cardiopatías Familiares. Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 3Hospital Infanta Cristina, Parla, Madrid, 4Servicio de Pediatría. Hospital General Universitario de Ciudad Real y 5Unidad de arritmias y electrofisiología. Servicio de Cardiología. Hospital General Universitario de Ciudad Real.
- 6055-4. CARACTERÍSTICAS CLÍNICAS DEL SÍNDROME DE QT LARGO INDUCIDO POR FÁRMACOS EN UNA GRAN COHORTE PROSPECTIVA
- Bieito Campos García, Héctor Hernández Ontiveros, Aina Ávila Parcet, Víctor García Hernando, Mar Carceller Sindreu, Juliana Salazar Blanco, Benjamín Rodríguez Santiago, Ana Juanes Borrego, Concepción Alonso Martín, Enrique Rodríguez Font, Zoraida L. Moreno Weidmann, Francisco Javier Méndez Zurita, Xavier Viñolas Prat y José M. Guerra Ramos
Hospital de la Santa Creu i Sant Pau, Barcelona.
- 6055-5. ESTUDIO DE LAS ALTERACIONES GENÉTICAS EN PACIENTES CON HIPERCOLESTEROLEMIA FAMILIAR DEL ÁREA SANITARIA DE TOLEDO, DETECTADOS MEDIANTE UNA NUEVA ESTRATEGIA DE CRIBADO SISTEMÁTICO PARTIENDO DE ANALÍTICA CENTRALIZADA PREEXISTENTE
- Joaquín Sánchez-Prieto Castillo, Esther Gigante Miravalles, Fernando Sabatel López, Alejandro Cabello Rodríguez, Carlos de Cabo Porras y Luis Rodríguez Padial
Hospital General Universitario de Toledo.
- 6055-6. GRADO DE CONTROL LIPÍDICO DE LOS PACIENTES CON HIPERCOLESTEROLEMIA FAMILIAR EN EL EMPORDÀ
- Patricia García Morante1, Daniel Vidal Soto1, Estel Pons Viñas1, Anna C. Comas Aleix2, Armand Grau Martín1 y Àlex Vila Belmonte1
1Hospital de Figueres, Girona y 2Fundació Salut Empordà, Figueres, Girona.
- 6055-7. PREDICTORES DE ENFERMEDAD CARDIOVASCULAR EN UNA UNIDAD DE HIPERCOLESTEROLEMIA FAMILIAR
- Gustavo Aníbal Cortez Quiroga, María Jesús Huertas Escribano, Elisa Martínez Perona, María de la Paz Eliche Mozas, Ana Cubillas Quero y Lara Cruz Romero
Hospital Alto Guadalquivir, Andújar (Jaén).
- 6055-8. DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- Yolanda Rico Ramírez1, Laura Torres-Juan1, Francisca Ramis Barceló2, Jaume Pons Llinares1, Elena Fortuny Frau1, Rafael Félix Ramis1, Víctor J. Asensio1, Icíar Martínez1, Vicente Peral Disdier1 y Damián Heine-Suñer1
1Hospital Son Espases, Palma de Mallorca y 2Clínica Planas, Palma de Mallorca.
- 6055-9. ANEURISMA DE AORTA ASCENDENTE GIGANTE COMO FORMA DE PRESENTACIÓN DE CUTIS LAXA 1B EN PACIENTES JÓVENES: NUEVO CASO Y REVISIÓN BIBLIOGRÁFICA
- Alejandro Used Gavín, José María Larrañaga Moreira, Esteban Martín Álvarez, Borja Souto Caínzos, Carmen Iglesias Gil, Víctor X. Mosquera Rodríguez, Roberto Barriales Villa y José Manuel Vázquez Rodríguez
Complexo Hospitalario Universitario A Coruña.
- 6055-10. ANÁLISIS DE LAS CARACTERÍSTICAS POBLACIONALES Y RENTABILIDAD DE LA IMPLANTACIÓN DE DAI EN PACIENTES CON MIOCARDIOPATÍA DILATADA NO ISQUÉMICA
- María Maeve Soto Pérez1, Jesús Piqueras Flores2, Javier Jiménez Díaz3, Felipe Higuera Sobrino3, Natalia Arance Romero4, Jorge Martínez del Río1, Martín Negreira Caamaño1, Daniel Águila Gordo1, Cristina Mateo Gómez1, Andrez Felipe Cubides Novoa1, Alfonso Morón Alguacil1, Pablo Soto Martín1, Emilio Blanco López1, Daniel Salas Bravo1 y José María Arizón Muñoz1
1Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 2Unidad de Cardiopatías Familiares. Servicio de Cardiología. Hospital General Universitario de Ciudad Real, 3Unidad de Arritmias y Electrofisiología. Servicio de Cardiología. Hospital General Universitario de Ciudad Real y 4Universidad de Castilla-La Mancha, Ciudad Real.
- 6055-11. RELEVANCIA DE LA TAQUICARDIA VENTRICULAR NO SOSTENIDA EN LA MIOCARDIOPATÍA HIPERTRÓFICA
- Charlotte Boillot, Belén Santos González, Ana Díaz Rojo, Andrea González Pigorini, Alejandro Cabello Rodríguez, Álvaro Serrano Blanco y Esther Gigante Miravalles
Complejo Hospitalario de Toledo, SESCAM.
Más comunicaciones de los autores
- Asensio, Víctor J.
- Félix Ramis, Rafael
-
Fortuny Frau, Elena
- 5014-8 - INCIDENCIA DE HEMORRAGIAS EN COMPLICACIONES VASCULARES TRAS IMPLANTE DE VÁLVULA AÓRTICA TRANSFEMORAL EN ANCIANOS
- 6033-8 - COMPLICACIONES VASCULARES MAYORES Y MENORES TRAS EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL: SIMILAR IMPACTO CLÍNICO, DIFERENTE PRONÓSTICO
- 6055-8 - DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- 6050-5 - USO DE LA IMAGEN AVANZADA EN EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL PARA EL ESTUDIO DE PREDICTORES DE COMPLICACIONES VASCULARES
- 6033-6 - IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL EN LA COMUNIDAD AUTÓNOMA DE LAS ISLAS BALEARES: ESTUDIO DE COHORTES SOBRE RESULTADOS Y COMPLICACIONES DE LOS ÚLTIMOS 10 AÑOS
- 6045-8 - COMPLICACIONES VASCULARES TRAS EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL: INCIDENCIA, PREDICTORES CLÍNICOS Y PRONÓSTICO
-
Heine Suñer, Damián
- 6055-8 - DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- 5031-5 - MIOCARDIOPATÍA HIPERTRÓFICA POR TRUNCAMIENTOS EN MYOSIN BINDING (MYBPC3): ¿QUÉ PODEMOS ESPERAR?
- 5031-2 - PRONÓSTICO DE LA MIOCARDIOPATÍA HIPERTRÓFICA POR TRUNCAMIENTO EN MYOSIN BINDING (MYBPC3): ¿SIRVEN LAS ESCALAS DE RIESGO CONVENCIONALES?
- Martínez, Icíar
-
Peral Disdier, Vicente
- 6055-8 - DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- 6045-8 - COMPLICACIONES VASCULARES TRAS EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL: INCIDENCIA, PREDICTORES CLÍNICOS Y PRONÓSTICO
- 6046-9 - PREVALENCIA Y EVOLUCIÓN DE LA DISLIPEMIA ATEROGÉNICA EN PACIENTES CON INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST: ESTUDIO DE COHORTES PROSPECTIVO
- 6033-6 - IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL EN LA COMUNIDAD AUTÓNOMA DE LAS ISLAS BALEARES: ESTUDIO DE COHORTES SOBRE RESULTADOS Y COMPLICACIONES DE LOS ÚLTIMOS 10 AÑOS
- 6026-9 - PAPEL DE LA DIMENSIÓN DEL LIGAMENTO DE MARSHALL EN EL ÉXITO DEL PROCEDIMIENTO DEL CIERRE PERCUTÁNEO DE LA OREJUELA IZQUIERDA
- 6009-6 - ¿ES EXTRAPOLABLE Y CONTEMPORÁNEA LA POBLACIÓN DE LOS ENSAYOS CLÍNICOS CITADOS EN LA GUÍA DE PRÁCTICA CLÍNICA DE LA SOCIEDAD EUROPEA DE CARDIOLOGÍA SOBRE INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL ST 2017?
- 6060-8 - ¿SE ALCANZAN LOS OBJETIVOS DE REDUCCIÓN DE C-LDL EN PACIENTES CON INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST?
- 4026-3 - BENEFICIOS OBSERVADOS DE ADMINISTRAR HEPARINA SÓDICA PRECOZMENTE EN LOS PACIENTES CON IAMCEST
- 6050-5 - USO DE LA IMAGEN AVANZADA EN EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL PARA EL ESTUDIO DE PREDICTORES DE COMPLICACIONES VASCULARES
- 5032-4 - INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST EN MENORCA: ¿IMPACTA REALMENTE LA GEOGRAFÍA Y LA ESTRATEGIA DE REPERFUSIÓN EN LA MORBIMORTALIDAD?
- 6047-8 - IMPACTO DEL TABACO SOBRE EL PRONÓSTICO TRAS UN INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST: UNA ASOCIACIÓN MEDIADA POR FACTORES DE CONFUSIÓN
- 6047-9 - INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL ST: ¿SE BASA LA GUÍA DE PRÁCTICA CLÍNICA DE LA ESC EN ENSAYOS CLÍNICOS DE PACIENTES CON IAMCEST?
- 6047-11 - EVALUACIÓN CRÍTICA DE LA EVIDENCIA PROPORCIONADA POR LAS RECOMENDACIONES DE LA GUÍA DE PRÁCTICA CLÍNICA DE LA ESC PARA EL MANEJO DEL INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST 2017
- 6002-5 - ¿QUÉ PARTICULARIDADES PRESENTAN LOS PACIENTES CON INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL ST QUE POSTERIORMENTE PRESENTAN UN EVENTO CEREBROVASCULAR EN EL SEGUIMIENTO?
- 6009-2 - DIFERENCIAS EN EL ESTADO DE COAGULACIÓN Y LA DOSIFICACIÓN DE HEPARINA SÓDICA ENTRE PACIENTES CON IAMCEST Y PACIENTES ESTABLES
- 6033-8 - COMPLICACIONES VASCULARES MAYORES Y MENORES TRAS EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL: SIMILAR IMPACTO CLÍNICO, DIFERENTE PRONÓSTICO
- 5014-8 - INCIDENCIA DE HEMORRAGIAS EN COMPLICACIONES VASCULARES TRAS IMPLANTE DE VÁLVULA AÓRTICA TRANSFEMORAL EN ANCIANOS
- 5007-1 - MODERADOR
-
Pons Llinares, Jaume
- 4024-2 - ESTUDIO VACCINE-CARDITIS: REGISTRO NACIONAL ESPAÑOL DE ENFERMEDAD INFLAMATORIA CARDIACA TRAS LA VACUNACIÓN CONTRA LA COVID-19
- 6002-5 - ¿QUÉ PARTICULARIDADES PRESENTAN LOS PACIENTES CON INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL ST QUE POSTERIORMENTE PRESENTAN UN EVENTO CEREBROVASCULAR EN EL SEGUIMIENTO?
- 5032-4 - INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST EN MENORCA: ¿IMPACTA REALMENTE LA GEOGRAFÍA Y LA ESTRATEGIA DE REPERFUSIÓN EN LA MORBIMORTALIDAD?
- 6055-8 - DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- 6047-8 - IMPACTO DEL TABACO SOBRE EL PRONÓSTICO TRAS UN INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST: UNA ASOCIACIÓN MEDIADA POR FACTORES DE CONFUSIÓN
-
Ramis Barceló, Francisca
- 4026-3 - BENEFICIOS OBSERVADOS DE ADMINISTRAR HEPARINA SÓDICA PRECOZMENTE EN LOS PACIENTES CON IAMCEST
- 6009-2 - DIFERENCIAS EN EL ESTADO DE COAGULACIÓN Y LA DOSIFICACIÓN DE HEPARINA SÓDICA ENTRE PACIENTES CON IAMCEST Y PACIENTES ESTABLES
- 6055-8 - DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
-
Rico Ramírez, Yolanda
- 4026-3 - BENEFICIOS OBSERVADOS DE ADMINISTRAR HEPARINA SÓDICA PRECOZMENTE EN LOS PACIENTES CON IAMCEST
- 6055-8 - DELECIÓN EN NOTCH 1, NUEVA CAUSA DE ANEURISMA AÓRTICO CON VÁLVULA AÓRTICA TRICÚSPIDE
- 6047-8 - IMPACTO DEL TABACO SOBRE EL PRONÓSTICO TRAS UN INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST: UNA ASOCIACIÓN MEDIADA POR FACTORES DE CONFUSIÓN
- 6009-2 - DIFERENCIAS EN EL ESTADO DE COAGULACIÓN Y LA DOSIFICACIÓN DE HEPARINA SÓDICA ENTRE PACIENTES CON IAMCEST Y PACIENTES ESTABLES
- 5014-8 - INCIDENCIA DE HEMORRAGIAS EN COMPLICACIONES VASCULARES TRAS IMPLANTE DE VÁLVULA AÓRTICA TRANSFEMORAL EN ANCIANOS
- 6033-8 - COMPLICACIONES VASCULARES MAYORES Y MENORES TRAS EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL: SIMILAR IMPACTO CLÍNICO, DIFERENTE PRONÓSTICO
- 6050-5 - USO DE LA IMAGEN AVANZADA EN EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL PARA EL ESTUDIO DE PREDICTORES DE COMPLICACIONES VASCULARES
- 6045-8 - COMPLICACIONES VASCULARES TRAS EL IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL: INCIDENCIA, PREDICTORES CLÍNICOS Y PRONÓSTICO
- 6026-9 - PAPEL DE LA DIMENSIÓN DEL LIGAMENTO DE MARSHALL EN EL ÉXITO DEL PROCEDIMIENTO DEL CIERRE PERCUTÁNEO DE LA OREJUELA IZQUIERDA
- 6002-5 - ¿QUÉ PARTICULARIDADES PRESENTAN LOS PACIENTES CON INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL ST QUE POSTERIORMENTE PRESENTAN UN EVENTO CEREBROVASCULAR EN EL SEGUIMIENTO?
- 6033-6 - IMPLANTE VALVULAR AÓRTICO TRANSFEMORAL EN LA COMUNIDAD AUTÓNOMA DE LAS ISLAS BALEARES: ESTUDIO DE COHORTES SOBRE RESULTADOS Y COMPLICACIONES DE LOS ÚLTIMOS 10 AÑOS
- 5032-4 - INFARTO AGUDO DE MIOCARDIO CON ELEVACIÓN DEL SEGMENTO ST EN MENORCA: ¿IMPACTA REALMENTE LA GEOGRAFÍA Y LA ESTRATEGIA DE REPERFUSIÓN EN LA MORBIMORTALIDAD?
- Torres-Juan, Laura