ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2020 - El e-Congreso de la Salud Cardiovascular

28 - 31 de Octubre de 2020
Introducción
Dr. Héctor Bueno
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
4013. Las mejores comunicaciones en innovación tecnológica y científica
Fecha
: 29-10-2020 10:30:00
Tipo
: Comunicaciones orales
Sala
: Sala 7
4013-3. IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
Antonio Manuel Lax Pérez1, Yassine Sassi2, Fernando Soler Pardo1, María Josefa Fernández del Palacio1, Álvaro Hernández Vicente1, David José Vázquez Andrés3, Eva Cabrera Romero3, David Fernández Vázquez3, Noelia Fernández Villa3, Manuel Veas Porlan3, Miguel Martínez Herrera3, Azucena Sáez Martín3, Antonio Escolar Conesa3, Domingo Andrés Pascual Figal3 y María del Carmen Asensio López4
1Universidad de Murcia. 2Mount Sinai Medical Center, Nueva York. 3Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 4Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia.
1Universidad de Murcia. 2Mount Sinai Medical Center, Nueva York. 3Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 4Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia.
Introducción y objetivos: Aunque varios estudios han relacionado la sobreexpresión de miR-199a-5p (miR199) con la disfunción cardiaca general tras infarto de miocardio (IM), los mecanismos subyacentes no se conocen. Recientemente, hemos sido pioneros en la identificación de Yin-yang 1 (Yy1) como un factor de transcripción capaz de regular la expresión de la isoforma sST2 bajo tensión biomecánica. Este estudio tiene como objetivo evaluar la implicación fisiopatológica de miR199a en la actividad cardiaca de Yy1 en relación con la expresión y liberación de sST2.
Métodos: Ratones C57BL/6 fueron sometidos a IM por ligadura permanente de la arteria descendente anterior izquierda. La administración de antimiR-control y antimiR199 fue intravenosa a 20 mg/kg; 24 horas después del IM. Las dimensiones y la función cardiaca se analizaron mediante ecocardiografía antes de la cirugía y tras el sacrificio (4 semanas tras el IM). Los análisis se llevaron a cabo en la zona remota del ventrículo izquierdo. La cuantificación del miR-199 así como los niveles de ARNm se evaluaron por PCR cuantitativa. La expresión de proteínas mediante RT-PCR y western blot. Se utilizó análisis in silico para predecir los objetivos plausibles de miR199 y ensayos de co-inmunoprecipitación para confirmar la interacción entre proteínas. Los niveles de sST2 se cuantificaron mediante ELISA.
Resultados: El IM se asoció con un aumento del nivel de miR-199, que promueve la hipertrofia de los miocitos cardiacos y la disfunción cardiaca general. En un modelo de ratón infartado, el silenciamiento sistémico de miR199, protegió significativamente de la hipertrofia y la fibrosis y mejoró la función cardiaca. Por análisis in silico, y posterior confirmación por ensayo de luciferasa, miR199 interacciona con Sirt1 e inhibe significativamente su expresión. El aumento de Sirt1 inducido por antimiR199 favoreció la interacción entre Sirt1 y la proteína pro-hipertrófica y co-activadora p300, lo que provocó su desacetilación e inhibición. Esta inhibición impidió la activación de Yy1, que bloqueó la expresión y la liberación de sST2.
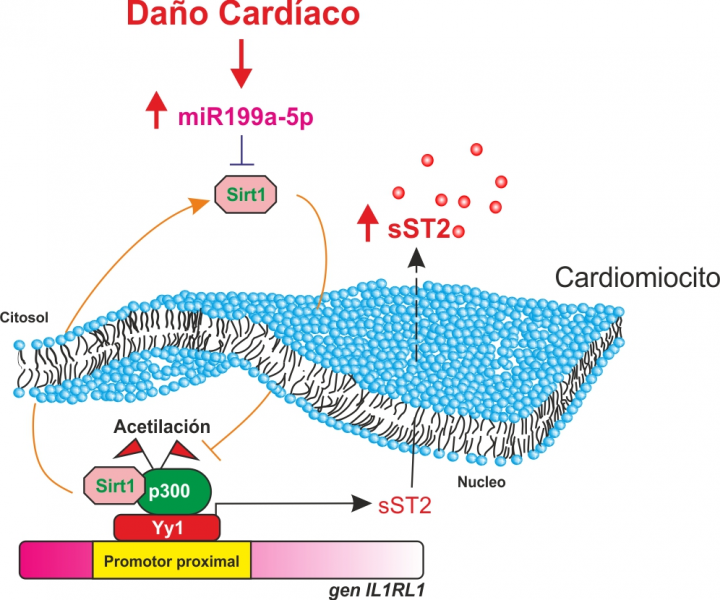
Mecanismo molecular de activación de Yy1 por miR199a.
Conclusiones: La super-regulación de Sirt1 inducida por la terapia con antimiR199 inactiva a p300, lo que provoca la inhibición de la actividad de Yy1 y, finalmente, una disminución de la expresión de sST2 después del IM. La manipulación de esta vía podría ofrecer nuevas opciones terapéuticas contra el remodelado adverso del miocardio.
Comunicaciones disponibles de "Las mejores comunicaciones en innovación tecnológica y científica"
- 4013-2. APRENDIZAJE AUTOMÁTICO (MACHINE LEARNING) EN EL DIAGNÓSTICO DEL INFARTO DE MIOCARDIO CON ARTERIAS CORONARIAS SIN ESTENOSIS SIGNIFICATIVAS. (MINOCA)
- María Jesús Espinosa Pascual1, Pablo Vaquero Martínez1, Víctor Vaquero Martínez2, Javier López Pais3, Barbara Izquierdo Coronel1, David Galán Gil1, Renée Olsen Rodríguez1, Rocío Abad Romero1 y Joaquín Jesús Alonso Martín1
1Hospital Universitario de Getafe (Madrid). 2Universidad de Valladolid. 3Centro Hospitalario Universitario de Santiago de Compostela (A Coruña).
- 4013-3. IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- Antonio Manuel Lax Pérez1, Yassine Sassi2, Fernando Soler Pardo1, María Josefa Fernández del Palacio1, Álvaro Hernández Vicente1, David José Vázquez Andrés3, Eva Cabrera Romero3, David Fernández Vázquez3, Noelia Fernández Villa3, Manuel Veas Porlan3, Miguel Martínez Herrera3, Azucena Sáez Martín3, Antonio Escolar Conesa3, Domingo Andrés Pascual Figal3 y María del Carmen Asensio López4
1Universidad de Murcia. 2Mount Sinai Medical Center, Nueva York. 3Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 4Instituto Murciano de Investigación Biosanitaria Virgen de la Arrixaca, Murcia.
- 4013-4. MONITORIZACIÓN NO INVASIVA Y NO IONIZANTE DE STENTS CORONARIOS MEDIANTE ESPECTROMETRÍA DE MICROONDAS
- Oriol Rodríguez-Leor1, Susana Amorós García de Valdecasas2, Giselle González-López2, Irene Jiménez2, Lluís Jofre2, Claudia Pérez-Martínez3, Antoni Bayes-Genis1, Javier Tejada Palacios4, Juan Manuel O'Callaghan Castellà2 y Carolina Gálvez Montón5
1Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona). 2Universidad Politécnica de Cataluña, Barcelona. 3Universidad de León. 4Universitat de Barcelona, Barcelona. 5Fundació Institut en Ciències de la Salut Germans Trias i Pujol, Badalona (Barcelona).
- 4013-5. EPIDEMIOLOGÍA DE LA ESTENOSIS AÓRTICA EN ESPAÑA. EVOLUCIÓN DE INGRESOS, CARACTERÍSTICAS DE LOS PACIENTES Y LETALIDAD HOSPITALARIA 2003-2015
- Nicolás Rosillo Ramírez1, Lourdes Vicent Alaminos2, David Martín de la Mota Sanz3, Francisco Javier Elola Somoza4 y Héctor Bueno5
1Servicio de Medicina Preventiva, Hospital Universitario 12 de Octubre, Madrid. 2Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid. 3Servicio de Medicina Preventiva y Gestión de Calidad, Hospital General Universitario Gregorio Marañon, Madrid. 4Fundación IMAS, Madrid. 5Servicio de Cardiología, Hospital Universitario 12 de Octubre, Madrid.
- 4013-6. NUCLEOPORINA 153, RAN GTP-ASA AP1, IMPORTINA 5 Y SERCA2A COMO POTENCIALES BIOMARCADORES DE RECHAZO CARDIACO DURANTE EL PRIMER AÑO TRAS EL TRASPLANTE CARDIACO
- Silvia Lozano Edo1, Ignacio Sánchez Lázaro1, Manolo Portolés2, Estefanía Tarazón3, Esther Roselló3, Meryem Ezzitouny1, Luis Almenar Bonet1, Raquel López Vilella1, Pablo Jover Pastor1 y Luis Martínez Dolz1
1Servicio de Cardiología del Hospital Universitario y Politécnico La Fe, Valencia. 2Instituto de Investigación Sanitaria del Hospital Universitario La Fe (IISLAFE), Valencia. 3Instituto de investigación Sanitaria del Hospital Universitario La Fe (IISLAFE), Valencia.
- 4013-7. CREACIÓN DE MODELOS PREDICTIVOS PARA LA PREDICCIÓN DE DESCOMPENSACIONES DE INSUFICIENCIA CARDIACA UTILIZANDO TÉCNICAS DE INTELIGENCIA ARTIFICIAL
- Vanessa Escolar Pérez1, Ainara Lozano Bahamonde1, Nekane Larburu Rubio2, Jon Kerexeta Sarriegi2, Arkaitz Artetxe2, Garazi Artola Balda2, Amaia Echebarria Chousa1 y Alberto Azkona Lucio1
1Hospital Universitario de Basurto, Bilbao (Vizcaya). 2Vicomtech, Donostia-San Sebastián (Guipúzcoa).
Más comunicaciones de los autores
-
Asensio López, María del Carmen
- 5020-13 - PREVALENCIA E IMPACTO PRONÓSTICO DE LAS MUTACIONES SOMÁTICAS EN PACIENTES CON INSUFICIENCIA CARDIACA Y FRACCIÓN DE EYECCIÓN REDUCIDA
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5013-8 - INFLUENCIA DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN EL PRONÓSTICO DE PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA
-
Cabrera Romero, Eva
- 6017-189 - VARIACIONES GEOGRÁFICAS EN LA PREVALENCIA DE CARDIOPATÍAS FAMILIARES Y EN LA INCIDENCIA DE MUERTE SÚBITA
- 5010-3 - FIABILIDAD INTRA E INTEROBSERVADOR ENTRE DOS MÉTODOS DE EVALUACIÓN DE STRAIN GLOBAL LONGITUDINAL DEL VENTRÍCULO IZQUIERDO. APLICACIONES DE LA INTELIGENCIA ARTIFICIAL
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 5016-14 - APLICACIÓN DEL NUEVO SCORE DE RIESGO DE MUERTE SÚBITA DE SIEIRA EN UNA POBLACIÓN CON SÍNDROME DE BRUGADA
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
-
Escolar Conesa, Antonio
- 6010-135 - IMPACTO DE LA CLASIFICACIÓN EHRA VALVULAR EN LOS PACIENTES CON FIBRILACIÓN AURICULAR
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5013-8 - INFLUENCIA DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN EL PRONÓSTICO DE PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA
- Fernández del Palacio, María Josefa
-
Fernández Vázquez, David
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 6020-201 - ECTASIA CORONARIA GRAVE FAMILIAR
- 5013-8 - INFLUENCIA DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN EL PRONÓSTICO DE PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA
- 5010-3 - FIABILIDAD INTRA E INTEROBSERVADOR ENTRE DOS MÉTODOS DE EVALUACIÓN DE STRAIN GLOBAL LONGITUDINAL DEL VENTRÍCULO IZQUIERDO. APLICACIONES DE LA INTELIGENCIA ARTIFICIAL
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
-
Fernández Villa, Noelia
- 5010-15 - INTELIGENCIA ARTIFICIAL EN LA EVALUACIÓN AUTOMÁTICA Y SEMIAUTOMÁTICA DEL STRAIN GLOBAL LONGITUDINAL PARA EL ANÁLISIS DE LA FUNCIÓN DEL VENTRÍCULO IZQUIERDO
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5010-3 - FIABILIDAD INTRA E INTEROBSERVADOR ENTRE DOS MÉTODOS DE EVALUACIÓN DE STRAIN GLOBAL LONGITUDINAL DEL VENTRÍCULO IZQUIERDO. APLICACIONES DE LA INTELIGENCIA ARTIFICIAL
-
Hernández Vicente, Álvaro
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
- 5013-8 - INFLUENCIA DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN EL PRONÓSTICO DE PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5020-13 - PREVALENCIA E IMPACTO PRONÓSTICO DE LAS MUTACIONES SOMÁTICAS EN PACIENTES CON INSUFICIENCIA CARDIACA Y FRACCIÓN DE EYECCIÓN REDUCIDA
-
Lax Pérez, Antonio Manuel
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
- 5020-13 - PREVALENCIA E IMPACTO PRONÓSTICO DE LAS MUTACIONES SOMÁTICAS EN PACIENTES CON INSUFICIENCIA CARDIACA Y FRACCIÓN DE EYECCIÓN REDUCIDA
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- Martínez Herrera, Miguel
-
Pascual Figal, Domingo Andrés
- 5013-8 - INFLUENCIA DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN EL PRONÓSTICO DE PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA
- 6015-170 - ANÁLISIS COMPARATIVO A 5 AÑOS DE LAS PRÓTESIS QUIRÚRGICAS SIN SUTURA FRENTE A PRÓTESIS TRANSCATÉTER EN UNA COHORTE DE PACIENTES REALES. ESTUDIO DE PROPENSIÓN DE 32 PAREJAS
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 6015-180 - FACTORES PREDICTORES DE REGRESIÓN DE MASA VENTRICULAR EN POSTOPERATORIO DE RECAMBIO VALVULAR AÓRTICO POR PRÓTESIS IMPLANTADAS DE MANERA QUIRÚRGICA FRENTE AL IMPLANTE TRANSCATÉTER
- 5017-10 - USO RACIONAL DE EXPLORACIONES COMPLEMENTARIAS EN ENFERMEDAD DE CHAGAS
- 5020-13 - PREVALENCIA E IMPACTO PRONÓSTICO DE LAS MUTACIONES SOMÁTICAS EN PACIENTES CON INSUFICIENCIA CARDIACA Y FRACCIÓN DE EYECCIÓN REDUCIDA
- 6046-390 - MALFORMACIÓN VASCULAR LINFÁTICO-QUÍSTICA PRIMARIA PERICÁRDICA COMO CAUSA RARA DE TAPONAMIENTO CARDIACO Y DISNEA PROGRESIVA
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
- 6062-469 - HIPOCAPNIA COMO FACTOR DE RIESGO PARA EL FRACASO DE LA VENTILACIÓN NO INVASIVA EN LA INSUFICIENCIA CARDIACA AGUDA
- 6035-349 - INSUFICIENCIA CARDIACA AGUDA DEBIDO A SÍNDROME CORONARIO AGUDO TRATADA CON VENTILACIÓN MECÁNICA NO INVASIVA. DIFERENCIAS ENTRE PACIENTES CON Y SIN ELEVACIÓN DEL SEGMENTO ST
- 6035-351 - DINÁMICA DEL POTASIO DURANTE LA HOSPITALIZACIÓN POR UN EPISODIO DE INSUFICIENCIA CARDIACA AGUDA
- 6074-544 - VALOR DE LDL INTRAHOSPITALARIO PARA PREDECIR LA CONSECUCIÓN DE NUEVOS OBJETIVOS DE LDL (MENOS DE 55 MG/DL) TRAS UN SÍNDROME CORONARIO AGUDO
- 6015-166 - ESTUDIO PRELIMINAR DEL IMPACTO ESTRUCTURAL DE LAS PRÓTESIS TRANSCATÉTER EN UNA COHORTE DE 179 PACIENTES REALES CONSECUTIVOS
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 6062-468 - EFECTIVIDAD Y SEGURIDAD DE LA VENTILACIÓN NO INVASIVA EN EL MANEJO DEL SHOCK CARDIOGÉNICO
-
Sáez Martín, Azucena
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5013-8 - INFLUENCIA DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN EL PRONÓSTICO DE PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA
- 6058-447 - ANÁLISIS DE LA FRACCIÓN DE EYECCIÓN DEL VENTRÍCULO IZQUIERDO EN PACIENTES CON PRIMERA HOSPITALIZACIÓN POR INSUFICIENCIA CARDIACA: PREVALENCIA, TENDENCIA TEMPORAL Y CARACTERÍSTICAS POBLACIONALES
- Sassi, Yassine
- Soler Pardo, Fernando
-
Vázquez Andrés, David José
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5010-3 - FIABILIDAD INTRA E INTEROBSERVADOR ENTRE DOS MÉTODOS DE EVALUACIÓN DE STRAIN GLOBAL LONGITUDINAL DEL VENTRÍCULO IZQUIERDO. APLICACIONES DE LA INTELIGENCIA ARTIFICIAL
- 5020-13 - PREVALENCIA E IMPACTO PRONÓSTICO DE LAS MUTACIONES SOMÁTICAS EN PACIENTES CON INSUFICIENCIA CARDIACA Y FRACCIÓN DE EYECCIÓN REDUCIDA
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO
-
Veas Porlan, Manuel
- 5003-12 - EMPAGLIFLOCINA MEJORA EL REMODEADO CARDIACO ADVERSO A TRAVÉS DE LA SOBRE-EXPRESIÓN DE LA ENZIMA GTP CICLOHIDROLASA 1
- 5010-3 - FIABILIDAD INTRA E INTEROBSERVADOR ENTRE DOS MÉTODOS DE EVALUACIÓN DE STRAIN GLOBAL LONGITUDINAL DEL VENTRÍCULO IZQUIERDO. APLICACIONES DE LA INTELIGENCIA ARTIFICIAL
- 4013-3 - IMPLICACIÓN DEL EJE DE SEÑALIZACIÓN MIRNA199A/SIRT1/P300/YY1/SST2 EN LA REGULACIÓN DEL REMODELADO ADVERSO TRAS EL INFARTO DE MIOCARDIO