ISSN: 0300-8932
Factor de impacto 2024
4,9
SEC 2020 - El e-Congreso de la Salud Cardiovascular

28 - 31 de Octubre de 2020
Introducción
Dr. Héctor Bueno
Presidente del Comité Científico del Congreso
Comité ejecutivo
Comité de evaluadores
Listado de sesiones
Índice de autores
4007. Miocardiopatías y amiloidosis cardiaca
Fecha
: 29-10-2020 09:30:00
Tipo
: Comunicaciones orales
Sala
: Sala 8
4007-2. CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
Fernando Domínguez Rodríguez1, Ángela López-Sainz2, Roberto Barriales-Villa3, Vicente Climent-Payá4, Coloma Tirón de Llano5, Mª Ángeles Espinosa Castro6, José Manuel García Pinilla7, Sergi César Díaz8, Enrique Santas Olmeda9, Esther Zorio Grima10, María Luisa Peña Peña11, Addison Julián Palomino Doza12, Perry Elliott13, Lorenzo Monserrat14 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 2Hospital Universitario Vall d'Hebron, Barcelona. 3Complexo Hospitalario Universitario A Coruña. 4Hospital General Universitario de Alicante. 5Hospital Universitario Dr. Josep Trueta, Girona. 6Hospital General Universitario Gregorio Marañón, Madrid. 7Hospital Clínico Universitario Virgen de la Victoria, Málaga. 8Hospital Sant Joan de Déu, Universitat de Barcelona. 9Hospital Clínico Universitario de Valencia. 10Hospital Universitario La Fe, Valencia. 11Hospital Universitario Virgen del Rocío, Sevilla. 12Hospital Universitario 12 de Octubre, Madrid. 13Barts Heart Centre, St Bartholomew's Hospital, Barts Health NHS Trust, London (Reino Unido). 14Health in Code, A Coruña.
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 2Hospital Universitario Vall d'Hebron, Barcelona. 3Complexo Hospitalario Universitario A Coruña. 4Hospital General Universitario de Alicante. 5Hospital Universitario Dr. Josep Trueta, Girona. 6Hospital General Universitario Gregorio Marañón, Madrid. 7Hospital Clínico Universitario Virgen de la Victoria, Málaga. 8Hospital Sant Joan de Déu, Universitat de Barcelona. 9Hospital Clínico Universitario de Valencia. 10Hospital Universitario La Fe, Valencia. 11Hospital Universitario Virgen del Rocío, Sevilla. 12Hospital Universitario 12 de Octubre, Madrid. 13Barts Heart Centre, St Bartholomew's Hospital, Barts Health NHS Trust, London (Reino Unido). 14Health in Code, A Coruña.
Introducción y objetivos: Variantes en el gen PRKAG2 pueden causar un síndrome caracterizado por miocardiopatía hipertrófica, trastornos de conducción y preexcitación ventricular. Hasta la fecha, solo se han reportado un número limitado de casos, y la historia natural de la enfermedad es poco conocida. El objetivo de este estudio es describir el fenotipo e historia natural de variantes en PRKAG2 en una cohorte extensa multicéntrica europea.
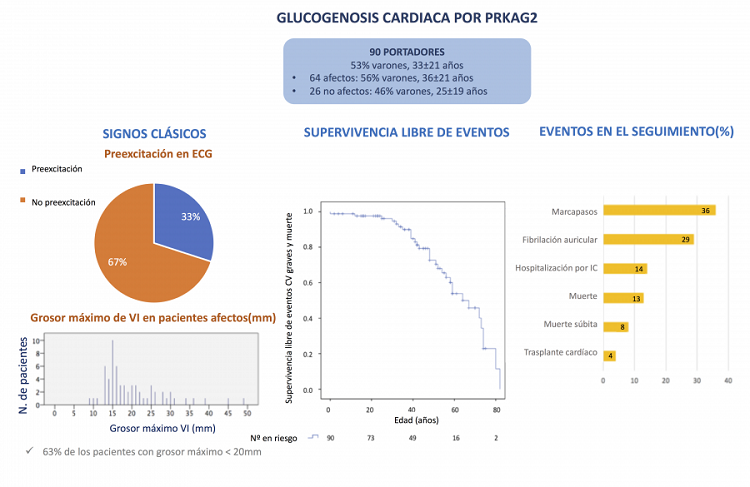
Métodos: Se incluyeron 90 pacientes portadores de variantes en PRKAG2 (53% varones, mediana de edad 33 años (RIQ: 15-50)) de 27 centros europeos. Se analizaron características clínicas, ECG y ecocardiograma en primera y última visita así como eventos clínicos en el seguimiento. Se incluyeron como eventos la fibrilación auricular (FA), eventos arrítmicos graves (taquicardia ventricular sostenida, fibrilación ventricular, choque apropiado de DAI y muerte súbita), implante de marcapasos, ingreso por insuficiencia cardiaca, trasplante cardiaco, implante de asistencia ventricular, muerte por causa cardiaca y muerte por cualquier causa.
Resultados: En la primera evaluación, 93% de los pacientes estaban en clase funcional NYHA I o II. El grosor máximo del VI era de 18 ± 8 mm y la fracción de eyección del VI (FEVI) de 61 ± 12%. Sesenta sujetos (67%) presentaban hipertrofia ventricular izquierda (HVI), 30 (33%) mostraban pre excitación en el ECG o se habían sometido a una ablación de vía accesoria, 17 (19%) portaban un marcapasos (edad media de implante 36 años (RIQ: 27-46)) y 16 (18%) tenían fibrilación auricular (FA) (edad media 43 años (RIQ: 31-54)). Después de una mediana de seguimiento de 6 años (RIQ: 2,3-13,9), el 71% presentaba HVI, el 29% FA y al 21% se le implantó un marcapasos de novo (mediana de edad al momento del implante 37 años (RIQ: 29-48)). Un 14% de los pacientes requirió ingreso por insuficiencia cardiaca (IC), 8% presentó muerte súbita o equivalente, 4% requirió un trasplante cardiaco y el 13% falleció.
|
Características clínicas en primera evaluación |
||
|
Portadores afectos (n = 64) |
Portadores no afectos (n = 26) |
|
|
Sexo masculino, n (%) |
36 (56) |
12 (46) |
|
Edad, años |
36 ± 21 |
25 ± 19 |
|
Historia familiar de muerte súbita, n (%) |
24 (38) |
11 (46) |
|
Ictus, n (%) |
4(6) |
0 |
|
Miopatía, n (%) |
2 (3) |
0 |
|
CK, U/L (rango) |
106 (2-365) |
66(2-130) |
|
NT-proBNP, pg/ml (mediana, RIQ) |
170 (37-2168) |
47 (10-224) |
|
Pre-excitación, n (%) |
30 (44) |
0 |
|
QRS, ms |
131 ± 37 |
108 ± 26 |
|
Fibrilación auricular, n (%) |
16 (25) |
0 |
|
Grosor máximo de VI, mm |
20 ± 8 |
10 ± 2 |
|
FEVI, % |
60 ± 13 |
66 ± 8 |
|
FEVI: fracción de eyección de ventrículo izquierdo; RIQ: rango intercuartílico; VI: ventrículo izquierdo. |
||
Fenotipo y pronóstico de cardiopatía por PRKAG2.
Conclusiones: La cardiopatía causada por variantes en PRKAG2 es una miocardiopatía progresiva caracterizada por alta incidencia de FA, trastornos de conducción, insuficiencia cardiaca avanzada y arritmias potencialmente mortales. Características clásicas como preexcitación y HVI grave no están presentes de manera uniforme y el diagnóstico debe considerarse en pacientes con HVI que desarrollan FA o requieren un marcapasos a edad temprana.
Comunicaciones disponibles de "Miocardiopatías y amiloidosis cardiaca"
- 4007-2. CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- Fernando Domínguez Rodríguez1, Ángela López-Sainz2, Roberto Barriales-Villa3, Vicente Climent-Payá4, Coloma Tirón de Llano5, Mª Ángeles Espinosa Castro6, José Manuel García Pinilla7, Sergi César Díaz8, Enrique Santas Olmeda9, Esther Zorio Grima10, María Luisa Peña Peña11, Addison Julián Palomino Doza12, Perry Elliott13, Lorenzo Monserrat14 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 2Hospital Universitario Vall d'Hebron, Barcelona. 3Complexo Hospitalario Universitario A Coruña. 4Hospital General Universitario de Alicante. 5Hospital Universitario Dr. Josep Trueta, Girona. 6Hospital General Universitario Gregorio Marañón, Madrid. 7Hospital Clínico Universitario Virgen de la Victoria, Málaga. 8Hospital Sant Joan de Déu, Universitat de Barcelona. 9Hospital Clínico Universitario de Valencia. 10Hospital Universitario La Fe, Valencia. 11Hospital Universitario Virgen del Rocío, Sevilla. 12Hospital Universitario 12 de Octubre, Madrid. 13Barts Heart Centre, St Bartholomew's Hospital, Barts Health NHS Trust, London (Reino Unido). 14Health in Code, A Coruña.
- 4007-3. CARACTERIZACIÓN DE LA CARDIOPATÍA AMILOIDÓTICA HEREDITARIA POR TRANSTIRETINA EN ESPAÑA
- Jorge Álvarez Rubio1, Ana José Manovel Sánchez2, Javier Limeres Freire3, José González Costello4, Pablo García-Pavía5, José Manuel García Pinilla6, Esther Zorio Grima7, María Valverde Gómez8, Mª Ángeles Espinosa Castro9, Gonzalo Barge Caballero10, Juan Ramón Gimeno Blanes11, Elena Fortuny Frau12, Luis Miguel Rincón Díaz13, María Gallego Delgado14 y Tomás Ripoll Vera1
1Hospital Hospital Son Llàtzer, Palma de Mallorca (Illes Balears). 2Hospital Juan Ramón Jiménez, Huelva. 3Hospital Universitario Vall d'Hebron, Barcelona. 4Hospital Universitario de Bellvitge, L'Hospitalet de Llobregat (Barcelona). 5Hospital Universitario Puerta de Hierro-Majadahonda (Madrid). 6Hospital Clínico Universitario Virgen de la Victoria, Málaga. 7Hospital Universitario La Fe, Valencia. 8Hospital Universitario 12 de Octubre, Madrid. 9Hospital General Universitario Gregorio Marañón, Madrid. 10Complexo Hospitalario Universitario A Coruña. 11Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 12Hospital Son Espases, Palma de Mallorca (Illes Balears). 13Hospital Universitario Ramón y Cajal, Madrid. 14Complejo Asistencial Universitario de Salamanca.
- 4007-4. VALIDACIÓN EXTERNA DEL SCORE PRONÓSTICO DE GILLMORE EN UNA COHORTE INTERNACIONAL DE PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTRETINA
- Fernando Domínguez Rodríguez1, Adrián Rivas Pérez1, Lindsey Mitrani2, Angelo Giuseppe Caponetti3, Roberta Mussinelli4, Esther González López1, Christian Gagliardi3, Ana Sabena4, Giulia Saturi3, Luis Enrique Escobar López1, Silvia Vilches Soria1, Claudio Rapezzi3, Mathew Maurer2 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 2Hospital Universitario de Columbia, Nueva York (New York). 3Universidad de Bolonia (Italia). 4Universidad de Pavía (Italia).
- 4007-5. ESTRATIFICACIÓN PRONÓSTICA EN NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- Guillem Casas Masnou1, Javier Limeres Freire1, Gerard Oristrell Santamaría1, Roberto Barriales Villa2, Juan Ramón Gimeno Blanes3, Pablo García Pavía4, Esther Zorio Grima5, Eduardo Villacorta Argüelles6, Juan Jiménez Jáimez7, Antoni Bayés-Genís8, José Manuel García Pinilla9, Addison Julián Palomino Doza10, Arturo Evangelista Masip1, Ignacio Ferreira González1 y José Fernando Rodríguez Palomares11
1Hospital Universitario Vall d'Hebron, Barcelona. 2Complexo Hospitalario Universitario A Coruña. 3Hospital Clínico Universitario Virgen de la Arrixaca, Murcia. 4Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 5Hospital Universitario La Fe, Valencia. 6Complejo Asistencial Universitario de Salamanca. 7Hospital Universitario Virgen de las Nieves, Granada. 8Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona). 9Hospital Clínico Universitario Virgen de la Victoria, Málaga. 10Hospital Universitario 12 de Octubre, Madrid. 11Hospital Universitario Vall d'Hebron, Barcelona, CIBERCV.
- 4007-6. FENOTIPO Y PRONÓSTICO DE LA MIOCARDIOPATÍA DILATADA EN PACIENTES PORTADORES DE MUTACIONES EN DISTROFINA
- María Alejandra Restrepo-Córdoba1, Juan Jiménez-Jáimez2, Vicente Climent-Payá3, Ana García-Álvarez4, José María Larrañaga5, Andrea Ros Peña6, Julián Palomino-Doza7, Luis Ruiz-Guerrero8, José F. Rodríguez-Palomares9, Coloma Tirón de Llano10, Mayte Basurte Elorz11, María Robledo Iñarritu12, Ramón Brugada10, Roberto Barriales-Villa5 y Pablo García-Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 2Hospital Universitario Virgen de las Nieves, Granada. 3Hospital General Universitario de Alicante. 4Hospital Clínic, Barcelona. 5Complexo Hospitalario Universitario A Coruña. 6Hospital Universitari Germans Trias i Pujol, Badalona (Barcelona). 7Hospital Universitario 12 de Octubre, Madrid. 8Hospital Universitario Marqués de Valdecilla, Santander (Cantabria). 9Hospital Universitario Vall d'Hebron, Barcelona. 10Hospital Universitario Dr. Josep Trueta, Girona. 11Complejo Hospitalario de Navarra, Pamplona (Navarra). 12Hospital Universitario Araba-Txagorritxu, Vitoria-Gasteiz (Álava).
- 4007-7. EVENTOS EMBÓLICOS EN AMILOIDOSIS CARDIACA POR TRANSTIRETINA. ANÁLISIS DE UNA COHORTE INTERNACIONAL
- Silvia Vilches Soria1, Esther González López1, Fernando Domínguez Rodríguez1, Luis Enrique Escobar López1, Adrián Rivas Pérez1, Aitor Hernández1, Stefano Perlini2, Claudio Rapezzi3, Mathew Maurer4, Julián Gillmore5 y Pablo García Pavía1
1Hospital Universitario Puerta de Hierro, Majadahonda (Madrid). 2Universidad de Pavía (Italia). 3Universidad de Bolonia (Italia). 4Hospital Universitario de Columbia, Nueva York (EEUU). 5Centro Nacional de Amiloidosis, Londres (Reino Unido).
Más comunicaciones de los autores
-
Barriales-Villa, Roberto
- 4007-5 - ESTRATIFICACIÓN PRONÓSTICA EN NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 5010-16 - VALOR DE UNA ECOCARDIOGRAFÍA DE EJERCICIO INTEGRAL PARA EL PRONÓSTICO DE PACIENTES CON MIOCARDIOPATÍA HIPERTRÓFICA
- 4007-6 - FENOTIPO Y PRONÓSTICO DE LA MIOCARDIOPATÍA DILATADA EN PACIENTES PORTADORES DE MUTACIONES EN DISTROFINA
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- César Díaz, Sergi
-
Climent-Payá, Vicente
- 6026-248 - MEJORÍA DE LOS NIVELES DE HEMOGLOBINA EN LOS PACIENTES CON ANEMIA SOMETIDOS A CIERRE DE OREJUELA
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 4007-6 - FENOTIPO Y PRONÓSTICO DE LA MIOCARDIOPATÍA DILATADA EN PACIENTES PORTADORES DE MUTACIONES EN DISTROFINA
-
Domínguez Rodríguez, Fernando
- 5019-12 - PREDICTORES DE DESARROLLO DE FIBRILACIÓN AURICULAR EN AMILOIDOSIS CARDIACA POR TRANSTIRETINA
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 5016-5 - PERFIL CLÍNICO Y PRONÓSTICO DE LA MIOCARDITIS Y MIOCARDIOPATÍA INFLAMATORIA CONFIRMADA POR BIOPSIA ENDOMIOCÁRDICA
- 4007-4 - VALIDACIÓN EXTERNA DEL SCORE PRONÓSTICO DE GILLMORE EN UNA COHORTE INTERNACIONAL DE PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTRETINA
- 5016-3 - SUPERIORIDAD DE LOS CRITERIOS INMUNOHISTOQUÍMICOS SOBRE LOS CRITERIOS DE DALLAS EN EL DIAGNÓSTICO DE MIOCARDITIS Y MIOCARDIOPATÍA INFLAMATORIA
- 5016-11 - EVENTOS EMBÓLICOS EN PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTIRETINA CON FIBRILACIÓN AURICULAR
- 4007-7 - EVENTOS EMBÓLICOS EN AMILOIDOSIS CARDIACA POR TRANSTIRETINA. ANÁLISIS DE UNA COHORTE INTERNACIONAL
- 5020-8 - EFICACIA Y SEGURIDAD DE LA FUROSEMIDA INTRAVENOSA JUNTO CON SUERO SALINO HIPERTÓNICO EN EL PACIENTE AMBULATORIO CON INSUFICIENCIA CARDIACA DESCOMPENSADA
- 4020-7 - PREVALENCIA E IMPACTO EN LA SUPERVIVENCIA DE LA FIBRILACIÓN AURICULAR EN PACIENTES CON AMILOIDOSIS CARDIACA TTR. ANÁLISIS DE UNA COHORTE INTERNACIONAL
- 6017-190 - EVOLUCIÓN DEL ACCESO VASCULAR PARA LA REALIZACIÓN DE BIOPSIA ENDOMIOCÁRDICA EN CORAZÓN NATIVO
- 5016-4 - EVENTOS EMBÓLICOS EN PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTIRETINA SIN FIBRILACIÓN AURICULAR
- 6005-27 - UTILIDAD DE LA BIOPSIA ENDOMIOCÁRDICA PARA EL DIAGNÓSTICO DE TUMORES CARDIACOS
- 6017-193 - FACTORES RELACIONADOS CON EL RETRASO DIAGNÓSTICO EN LA AMILOIDOSIS CARDIACA POR TRANSTIRRETINA
- Elliott, Perry
-
Espinosa Castro, María Ángeles
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 4007-3 - CARACTERIZACIÓN DE LA CARDIOPATÍA AMILOIDÓTICA HEREDITARIA POR TRANSTIRETINA EN ESPAÑA
- 6012-142 - POTENCIAL IMPACTO DE VARIANTES MISSENSE Y SINÓNIMAS EN TITINA EN UNA COHORTE DE PACIENTES CON MIOCARDIOPATÍA DILATADA
-
García Pavía, Pablo
- 5020-8 - EFICACIA Y SEGURIDAD DE LA FUROSEMIDA INTRAVENOSA JUNTO CON SUERO SALINO HIPERTÓNICO EN EL PACIENTE AMBULATORIO CON INSUFICIENCIA CARDIACA DESCOMPENSADA
- 4007-6 - FENOTIPO Y PRONÓSTICO DE LA MIOCARDIOPATÍA DILATADA EN PACIENTES PORTADORES DE MUTACIONES EN DISTROFINA
- 4007-4 - VALIDACIÓN EXTERNA DEL SCORE PRONÓSTICO DE GILLMORE EN UNA COHORTE INTERNACIONAL DE PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTRETINA
- 6017-193 - FACTORES RELACIONADOS CON EL RETRASO DIAGNÓSTICO EN LA AMILOIDOSIS CARDIACA POR TRANSTIRRETINA
- 6017-190 - EVOLUCIÓN DEL ACCESO VASCULAR PARA LA REALIZACIÓN DE BIOPSIA ENDOMIOCÁRDICA EN CORAZÓN NATIVO
- 4020-7 - PREVALENCIA E IMPACTO EN LA SUPERVIVENCIA DE LA FIBRILACIÓN AURICULAR EN PACIENTES CON AMILOIDOSIS CARDIACA TTR. ANÁLISIS DE UNA COHORTE INTERNACIONAL
- 4007-7 - EVENTOS EMBÓLICOS EN AMILOIDOSIS CARDIACA POR TRANSTIRETINA. ANÁLISIS DE UNA COHORTE INTERNACIONAL
- 5019-12 - PREDICTORES DE DESARROLLO DE FIBRILACIÓN AURICULAR EN AMILOIDOSIS CARDIACA POR TRANSTIRETINA
- 4007-5 - ESTRATIFICACIÓN PRONÓSTICA EN NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 4007-3 - CARACTERIZACIÓN DE LA CARDIOPATÍA AMILOIDÓTICA HEREDITARIA POR TRANSTIRETINA EN ESPAÑA
- 5016-5 - PERFIL CLÍNICO Y PRONÓSTICO DE LA MIOCARDITIS Y MIOCARDIOPATÍA INFLAMATORIA CONFIRMADA POR BIOPSIA ENDOMIOCÁRDICA
- 5016-11 - EVENTOS EMBÓLICOS EN PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTIRETINA CON FIBRILACIÓN AURICULAR
- 5016-4 - EVENTOS EMBÓLICOS EN PACIENTES CON AMILOIDOSIS CARDIACA POR TRANSTIRETINA SIN FIBRILACIÓN AURICULAR
- 5016-3 - SUPERIORIDAD DE LOS CRITERIOS INMUNOHISTOQUÍMICOS SOBRE LOS CRITERIOS DE DALLAS EN EL DIAGNÓSTICO DE MIOCARDITIS Y MIOCARDIOPATÍA INFLAMATORIA
- 5010-2 - VALOR DEL ECOCARDIOGRAMA DE ESFUERZO EN EL PRONÓSTICO DE LA AMILOIDOSIS CARDIACA SENIL
- 6005-27 - UTILIDAD DE LA BIOPSIA ENDOMIOCÁRDICA PARA EL DIAGNÓSTICO DE TUMORES CARDIACOS
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
-
García Pinilla, José Manuel
- 6083-621 - CARACTERÍSTICAS DE LOS PACIENTES CON INSUFICIENCIA CARDIACA Y FIBRILACIÓN AURICULAR TRATADOS CON RIVAROXABÁN. DATOS DEL ESTUDIO FARAONIC
- 5005-7 - CARACTERÍSTICAS CLÍNICAS Y ANALÍTICAS DE LOS PACIENTES MAYORES CON IC EN RELACIÓN CON SU FUNCIÓN VENTRICULAR
- 4007-3 - CARACTERIZACIÓN DE LA CARDIOPATÍA AMILOIDÓTICA HEREDITARIA POR TRANSTIRETINA EN ESPAÑA
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 6011-140 - PREVALENCIA DE LOS FACTORES DE RIESGO CARDIOVASCULAR EN PACIENTES MAYORES CON INSUFICIENCIA CARDIACA AMBULATORIA SEGUIDOS POR CARDIOLOGÍA EN ESPAÑA
- 4007-5 - ESTRATIFICACIÓN PRONÓSTICA EN NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 4018-4 - PREVALENCIA Y CARACTERÍSTICAS DE LOS SÍNDROMES GERIÁTRICOS EN PACIENTES MAYORES CON INSUFICIENCIA CARDIACA SEGÚN SU FUNCIÓN VENTRICULAR
-
López Sainz, Ángela
- 4019-6 - PREDICTORES DE MORTALIDAD EN EL SÍNDROME AÓRTICO AGUDO TIPO A Y TIPO B: ANÁLISIS DEL REGISTRO ESPAÑOL DEL SÍNDROME AÓRTICO AGUDO (RESA)
- 4019-4 - TRATAMIENTO QUIRÚRGICO DE LA DISECCIÓN TIPO A EN EL REGISTRO ESPAÑOL DEL SÍNDROME AÓRTICO AGUDO. UNA MEJORA CONTINUADA
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 4019-2 - REGISTRO ESPAÑOL DEL SÍNDROME AÓRTICO AGUDO (RESA- III). LOS CAMBIOS EN EL MANEJO TERAPÉUTICO SE REFLEJAN EN UNA DISMINUCIÓN SIGNIFICATIVA DE LA MORTALIDAD
- 6043-387 - ¿SON LOS DIÁMETROS ECOCARDIOGRÁFICOS DE LA RAÍZ AÓRTICA Y AORTA ASCENDENTE INTERCAMBIABLES SEGÚN LAS GUÍAS PEDIÁTRICAS VS LAS DE ADULTOS?
- Monserrat, Lorenzo
-
Palomino Doza, Addison Julián
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 4007-5 - ESTRATIFICACIÓN PRONÓSTICA EN NO COMPACTACIÓN DEL VENTRÍCULO IZQUIERDO
- 4007-6 - FENOTIPO Y PRONÓSTICO DE LA MIOCARDIOPATÍA DILATADA EN PACIENTES PORTADORES DE MUTACIONES EN DISTROFINA
- 6060-457 - PERFIL GENÉTICO-MOLECULAR DE UNA COHORTE ESPAÑOLA DE HIPERTENSIÓN ARTERIAL PULMONAR ASOCIADA A ENFERMEDADES DE TEJIDO CONECTIVO
- 5013-6 - DIAGNÓSTICO MEDIANTE PANEL DE SECUENCIACIÓN MASIVA EN UNA COHORTE NACIONAL DE HIPERTENSIÓN ARTERIAL PULMONAR
- 5013-13 - PATRONES ERGOESPIROMÉTRICOS EN PACIENTES CON HIPERTENSIÓN ARTERIAL PULMONAR: ¿PODEMOS SOSPECHAR ENFERMEDAD VENO-OCLUSIVA?
- Peña Peña, María Luisa
-
Santas Olmeda, Enrique
- 4015-2 - FUNCIÓN SISTÓLICA VENTRICULAR IZQUIERDA Y DERECHA TRAS LA ADMINISTRACIÓN DE HIERRO CARBOXIMALTOSA: ANÁLISIS POST-HOC DE RMC DEL ESTUDIO MYOCARDIAL-IRON
- 4009-5 - SST2 Y EFICIENCIA DIURÉTICA EN INSUFICIENCIA RENAL AGUDA Y DISFUNCIÓN RENAL CONCOMITANTE
- 5005-8 - PERFIL CLÍNICO Y PRONÓSTICO DE LOS PACIENTES MUY ANCIANOS CON INSUFICIENCIA CARDIACA AGUDA
- 4006-6 - RIESGO DE REHOSPITALIZACIONES POR INSUFICIENCIA CARDIACA EN BASE A LA COMBINACIÓN DE PARÁMETROS DERECHOS EN LA INSUFICIENCIA CARDIACA CON FUNCIÓN SISTÓLICA CONSERVADA
- 5020-2 - DINÁMICA DE LA FUNCIÓN RENAL TRAS LA ADMINISTRACIÓN DE SACUBITRILO/VALSARTÁN Y EMPAGLIFOZINA EN PACIENTES CON INSUFICIENCIA CARDIACA Y DIABETES TIPO 2
- 4009-2 - NIVELES MÁS BAJOS DE CA125 SE RELACIONAN CON UN MEJOR PRONÓSTICO A CORTO PLAZO EN PACIENTES CON INSUFICIENCIA CARDIACA AGUDA
- 4007-2 - CARACTERÍSTICAS CLÍNICAS E HISTORIA NATURAL DE LA GLUCOGENOSIS CARDIACA POR VARIANTES EN PRKAG2
- 6043-381 - CRITERIOS ECOCARDIOGRÁFICOS ESTRICTOS DE NEGATIVIDAD Y PREDICTORES DE ECOCARDIOGRAFÍAS DE SEGUIMIENTO EN LA SOSPECHA DE ENDOCARDITIS INFECCIOSA
- 6067-505 - FUNCIÓN SISTÓLICA VENTRICULAR IZQUIERDA Y DERECHA Y DÉFICIT DE HIERRO EN INSUFICIENCIA CARDIACA AGUDA
- 6060-455 - VALOR PRONÓSTICO DEL DIÁMETRO INDEXADO DE LA ARTERIA PULMONAR OBTENIDO MEDIANTE RESONANCIA MAGNÉTICA CARDIACA EN PACIENTES CON INSUFICIENCIA CARDIACA AGUDA
- Tirón de Llano, Coloma
- Zorio Grima, Esther