Sra. Editora:
Presentamos una familia con cuatro miembros diagnosticados molecularmente de síndrome de Marfan (MFS), con una expresión muy variable. El caso índice es un varón de 30 años al que se detectó a los 12 una dilatación leve de la raíz aórtica. Fue intervenido de cataratas a los 13 y a los 18 apareció una subluxación de cristalino, y entonces se le diagnosticó MFS. Actualmente presenta ceguera total del ojo derecho, escoliosis, aracnodactilia y luxación rotuliana recidivante. El diámetro de la raíz aórtica actualmente es de 41 mm.
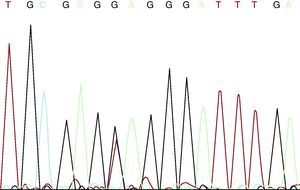
El paciente y su pareja habían acudido a la unidad de genética solicitando consejo genético para futura descendencia. Él refería tener cuatro hermanas sanas y que él era el único afectado de MFS en su familia, sin antecedentes familiares de enfermedad aórtica o muerte súbita a edad joven. Tras explicar el tipo de herencia —autosómica dominante— y barajar opciones reproductivas, se ofreció la posibilidad de diagnóstico prenatal o diagnóstico preimplantacional una vez que se conociera la mutación causante de su enfermedad, y se les informó de que más del 90% de los pacientes con MFS pueden presentar una mutación en el gen FBN1. Se discutió con la pareja las limitaciones en la predicción de la gravedad de la enfermedad, dado que el espectro clínico varía desde la afección osteoarticular leve hasta formas graves neonatales con enfermedad cardiovascular mortal. Tras el adecuado consentimiento informado, se procedió a secuenciar el gen FBN1 a partir de ADN extraído de sangre periférica. En dicho estudio se detectaron cuatro polimorfismos de un solo nucleótido (SNP) incluidos en la SNP database (en Entrez databases): rs1018148 (IVS2–102T>C), rs59966849 (IVS28+47–/CATAA), rs2303502 (IVS48+54T>A) y rs363832 (IVS56+17G>C). Además, se detectaron dos variantes intrónicas no descritas en las bases de datos de polimorfismos y, por lo tanto, de significado incierto (IVS25+49delTAAAGA y IVS40–35C>T). En las secuencias codificantes se identificó una mutación puntual en heterocigosis en el exón 54: E2194X (c.6580G>T) (Figura 1). Se trata de una mutación de terminación de codón prematura (como el 33% de las mutaciones de este gen) no descrita hasta el momento, presumible causa de la enfermedad del paciente por el truncamiento y la pérdida del 24% de la fibrilina 1, que afecta a 11 dominios EGF-like (epidermal growth factor like). Sería una mutación nonsense potencialmente patogénica según los criterios revisados de Gante. La mutación está localizada en el exón 54, en la región 3’, donde se ha descrito el 37% de las mutaciones de FBN1.

Figura 1. Mutación c.6580G>T.
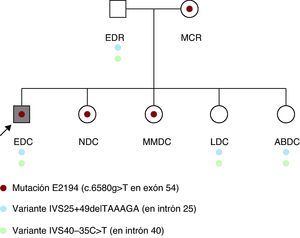
Inicialmente se estudió la presencia de la mutación y de las dos variantes inciertas en los padres, y se detectó inesperadamente en su madre, aparentemente sana, la misma mutación. En el padre, de 53 años y 169 cm de estatura, con exploración física, electrocardiograma y ecocardiograma normal, se detectaron las dos variantes de significado incierto. La exploración de la madre, de 50 años y 160 cm de estatura, reveló una cifoescoliosis pronunciada, hipotonía abdominal y una ectasia del saco dural por resonancia magnética (RM). El examen oftalmológico y el ecocardiograma transtorácico resultaron normales. Posteriormente se realizó estudio molecular y exploración física a las cuatro hermanas. Se detectó en dos de ellas (NDC y MMDC) la misma mutación patogénica y en las otras dos (LDC y ABDC), sólo las dos variantes de significado incierto (Figura 2). En la exploración de NDC, de 23 años y 172 cm, se detectó pectus carinatum, aracnodactilia, signos positivos del pulgar y de la muñeca e hiperlaxitud marcada. La RM mostró una escoliosis leve y a nivel lumbosacro una ectasia del saco dural. El examen oftalmológico y el ecocardiograma fueron normales. Sus otras tres hermanas sanas rechazaron la exploración, aunque MMDC, portadora de la mutación, tiene escoliosis.

Figura 2. Árbol familiar.
El análisis molecular nos permitió identificar de forma inesperada a tres familiares sanos que habían estado infradiagnosticados durante años.
Los hallazgos en esta familia nos permiten sacar varias conclusiones: a) el estudio genético debe comenzarse con el paciente más afectado y, en caso de detectarse mutación causal, proseguir con familiares; b) teniendo en cuenta la morbimortalidad de los pacientes con MFS, resulta importante su diagnóstico precoz para un adecuado manejo médico y quirúrgico1, para lo cual resultan efectivos los consensos internacionales de criterios diagnósticos revisados recientemente2, y en casos de expresión mínima, el diagnóstico genético puede no alterar demasiado el manejo clínico, pero sí permite opciones reproductivas y ayuda en la evaluación del resto de los familiares3; c) el estudio molecular en familiares, a priori con riesgo del 50% de estar afectados, permite identificar a aquellos con predisposición a padecer la enfermedad y conlleva además un ahorro en revisiones periódicas en caso de que no tengan la mutación; d) la RM permite la identificación de ectasia dural, presente en el 90% de los pacientes MFS, con alta especificidad diagnóstica, como se ha apuntado en la revisión de los criterios Gante2; e) se debe tener especial cuidado en la transmisión de la información, pues en algunos casos la situación es similar a la de los análisis predictivos; f) respecto a la variabilidad en la expresión, Van Dijk et al4 señalan que mutaciones adicionales en trans en FBN1 podrían tener un papel modificador que explique la variabilidad intrafamiliar; nuestros hallazgos podrían reafirmar esta hipótesis, ya que se han detectado dos variantes que cuando acompañan a la mutación el fenotipo es más severo y pueden tener un papel modificador del fenotipo; g) como han apuntado Comeglio et al5, es primordial para la comunidad científica identificar el mayor número de mutaciones para tener más claros los efectos causales o modificadores de mutaciones similares, lo que ayudaría al consejo genético, definir el probable pronóstico y controlar decisiones en el manejo de los pacientes, y h) en caso de no disponer de estudios genéticos, es obligada la valoración clínica de los familiares tras un diagnóstico de cardiopatía hereditaria en un probando.
Autor para correspondencia: adiazb.hmtl@salud.madrid.org