To the Editor,
We present a family in which four members were diagnosed by molecular testing as having the Marfan syndrome (MFS), with a highly variable expression. The proband is a 30-year-old man in whom a mild dilatation of the aortic root was detected at the age of 12 years. He underwent cataract surgery when he was 13 and, when he was found to have lens subluxation at the age of 18, he was diagnosed with MFS. At the present time, he is totally blind in his right eye and has scoliosis, arachnodactyly and recurrent patellar dislocation. The diameter of the aortic root is presently 41mm.
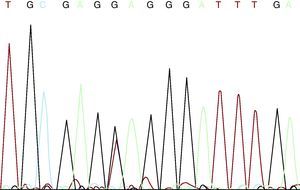
The patient and his partner came to the genetics unit seeking genetic counseling with regard to future offspring. He explained that he had four healthy sisters, that he was the only member of his family with MFS, and that there was no family history of aortic disease or sudden death at a young age. After receiving an explanation of the mode of inheritance—autosomal dominant—and a description of the reproductive options, they were offered prenatal or preimplantation diagnosis once the mutation responsible for the disease had been identified. They were also informed that more than 90% of the patients with MFS may have an FBN1 gene mutation. We discussed with the couple the limitations to predict the severity of the disease, given that the clinical spectrum ranges from mild bone and joint involvement to severe neonatal forms with fatal cardiovascular disease. After adequate informed consent, we proceeded to FBN1 gene sequencing using DNA extracted from peripheral blood. In this study, we detected four single nucleotide polymorphisms (SNP) included in the SNP database (in Entrez databases): rs1018148 (IVS2–102T>C), rs59966849 (IVS28+47–/CATAA), rs2303502 (IVS48+54T>A) and rs363832 (IVS56+17G>C). In addition, we detected two intronic variants not described in the polymorphism databases and, thus, of uncertain significance (IVS25+49delTAAAGA and IVS40–35C>T). In the coding sequences, we identified a heterozygous point mutation in exon 54: E2194X (c.6580G >T) (Figure 1). It was a premature termination codon (as are 33% of the mutations of this gene) that had not been previously described. This mutation presumably causes the disease as it produces the truncation and loss of 24% of the fibrillin 1, which affects 11 epidermal growth factor-like domains. It would be a potentially pathogenic nonsense mutation, according to the revised Ghent criteria. The mutation is located in exon 54, in the 3’ region, where 37% of the mutations of FBN1 have been described.

Figure 1. c.6580G >T mutation.
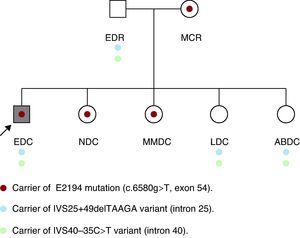
Initially, we studied the presence of the mutation and of the two uncertain variants in the parents of the patient, with the unexpected detection of the same mutation in his mother, who was apparently healthy. In his father (age: 53 years; height: 169cm), who had a normal physical examination, electrocardiogram and echocardiogram, we detected two variants of uncertain significance. The examination of his mother (age: 50 years; height: 160cm) revealed marked kyphoscoliosis, abdominal hypotonia and dural ectasia in the magnetic resonance (MR) study. The results of the ophthalmologic examination and transthoracic echocardiogram were normal. Subsequently, his four sisters underwent a molecular study and physical examination. In two of them (NDC and MMDC), the same pathogenic mutation was detected, and in the other two (LDC and ABDC), only the two variants of uncertain significance were observed (Figure 2). In the examination of NDC (age: 23 years; height: 172cm), we detected pectus carinatum, arachnodactyly, positive thumb and wrist signs and marked hyperlaxity. Magnetic resonance imaging demonstrated mild scoliosis, as well as dural ectasia at the lumbosacral level. The results of the ophthalmologic examination and echocardiogram were normal. The other three healthy sisters refused to undergo the examination, although MMDC, a carrier of the mutation, had scoliosis.

Figure 2. Family tree.
Unexpectedly, the molecular analysis enabled us to identify three healthy relatives in whom the correct diagnosis had been overlooked for years.
The findings in this family lead us to several conclusions: a) the genetic study should commence with the most severely affected patients and, if a causative mutation is detected, should be continued with relatives; b) taking into account the morbidity and mortality rates in patients with MFS, early diagnosis is important so that appropriate medical and surgical management can be undertaken;1 the recently revised international consensus guidelines for diagnostic criteria2 are valuable for this purpose and, in cases of minimal expression, the genetic diagnosis may not change the clinical management a great deal, but it will permit reproductive options and aid in the evaluation of other relatives;3 c) the molecular study in relatives, a priori with a 50% risk of being affected, makes it possible to identify those with a predisposition to the disease and, moreover, results in a savings in periodical checkups in the case they do not have the mutation; d) MR enables the identification of dural ectasia, present in 90% of the patients with MFS, with a high diagnostic specificity, as has been pointed out in the revision of the Ghent criteria;2 e) special care should be taken in the transmission of the information since, in some cases, the situation is similar to that of the predictive analyses; f) with respect to the variability in expression, Van Dijk et al.4 pointed out that additional trans-acting mutations in FBN1 could play a role as modifiers that would explain the intrafamilial variability; our findings could strengthen this hypothesis since we detected two variants that, when they accompany the mutation, are associated with a more severe phenotype, and that may play a role as modifiers of the phenotype; g) as Comeglio et al. have pointed out,5 it is essential that the scientific community identify the largest possible number of mutations in order to further clarify the causative or modifying effects of similar mutations, a circumstance that would aid in genetic counseling, to define the probable diagnosis and to control decisions made in patient management; and h) if genetic studies are not available, clinical evaluation of relatives is indispensable after a diagnosis of hereditary heart disease in a proband.
.
Corresponding author: adiazb.hmtl@salud.madrid.org