






























tridimensional
aurícula derecha
aurícula izquierda
coronaria izquierda anómala desde la arteria pulmonar
arteria pulmonar
antagonistas del receptor de la angiotensina II
coronaria derecha anómala desde la arteria pulmonar
antagonistas del receptor de la endotelina
auriculoventricular
área valvular aórtica
área valvular aórtica indexada
péptido natriurético cerebral
coronaria anómala con origen en la arteria pulmonar
cirugía de revascularización coronaria
cardiopatía congénita
cardiopatía congénita del adulto
comunicación interauricular
comunicación interventricular
coartación aórtica
corazón univentricular
desfibrilador automático implantable
ductus arterioso persistente
derecho-izquierdo
defecto del septo auriculoventricular
defecto del septo ventricular
diámetro telesistólico del ventrículo izquierdo
estenosis aórtica
European Association of Cardiovascular Imaging
estenosis aórtica supravalvular
enfermedad coronaria
enzima de conversión de la angiotensina
electrocardiograma
estimulación eléctrica programada
electrofisiológico
enfermedad hereditaria de la aorta torácica
endocarditis infecciosa
estenosis pulmonar
estenosis subaórtica
Sociedad Europea de Cardiología
ecocardiografía transesofágica
ecocardiografía transtorácica
enfermedad vascular pulmonar
fibrilación auricular
fosfodiesterasa 5
fracción de eyección
fracción de eyección del ventrículo derecho
fracción de eyección del ventrículo izquierdo
fibrilación ventricular
hipertensión arterial pulmonar
hipertensión arterial pulmonar asociada con cardiopatía congénita
hipertensión pulmonar
hipertrofia ventricular derecha
hipertrofia ventricular izquierda
insuficiencia aórtica
izquierdo-derecho
razón internacional normalizada
insuficiencia pulmonar
implante percutáneo de válvula pulmonar
insuficiencia tricuspídea
índice de tamaño aórtico
colaterales aortopulmonares principales
marcapasos
muerte súbita cardiaca
nuevos anticoagulantes orales no antagonistas de la vitamina K
fragmento aminoterminal del propéptido natriurético cerebral
New York Heart Association
origen aórtico anómalo de una arteria coronaria
origen aórtico anómalo de la coronaria derecha
origen aórtico anómalo de la coronaria izquierda
Organización Mundial de la Salud
obstrucción del tracto de salida del ventrículo derecho
obstrucción del tracto de salida del ventrículo izquierdo
presión arterial pulmonar
prueba de ejercicio cardiopulmonar
prueba de los 6 min de marcha
presión ventricular derecha
cociente de flujo pulmonar a sistémico
resonancia magnética cardiovascular
resistencia vascular pulmonar
síndrome del hemicardio izquierdo hipoplásico
sustitución de válvula pulmonar
taquicardia auricular
taquicardia auricular ectópica
tomografía computarizada cardiovascular
tetralogía de Fallot
tetralogía de Fallot reparada
transposición de las grandes arterias
transposición de las grandes arterias corregida congénitamente
taquicardia por reentrada auriculoventricular
terapia de resincronización cardiaca
taquicardia por reentrada intraauricular
taquicardia por reentrada del nódulo auriculoventricular
taquicardia supraventricular
tracto de salida del ventrículo derecho
tracto de salida del ventrículo izquierdo
taquicardia ventricular
unidades Wood
válvula aórtica bicúspide
vena cava inferior
volumen corpuscular medio
vena cava superior
ventrículo derecho
ventrículo derecho con doble cámara
pendiente entre ventilación pulmonar (VE) y producción de CO2
ventrículo izquierdo
velocidad Doppler máxima
válvula tricúspide
volumen telediastólico del ventrículo derecho indexado
volumen telesistólico del ventrículo derecho indexado
TABLA DE CONTENIDOS
1. Preámbulo
2. Introducción
2.1. ¿Por qué se necesita una guía nueva sobre el tratamiento de las cardiopatías congénitas del adulto?
2.2. Contenido de la guía
2.3. Nuevo formato de la guía
2.4. Cómo usar esta guía
2.5. Qué hay de nuevo en la guía de 2020
3. Aspectos generales
3.1. Prevalencia de las cardiopatías congénitas del adulto
3.2. Organización de la atención
3.3. Estudio diagnóstico
3.3.1. Ecocardiografía
3.3.2. Resonancia magnética cardiovascular
3.3.3. Tomografía computarizada cardiovascular
3.3.4. Pruebas de esfuerzo cardiopulmonar
3.3.5. Cateterismo cardiaco
3.3.6. Biomarcadores
3.4. Consideraciones terapéuticas
3.4.1. Insuficiencia cardiaca
3.4.2. Arritmias y muerte súbita cardiaca
3.4.2.1. Sustratos arritmogénicos
3.4.2.2. Evaluación de pacientes con arritmia sospechada o documentada y tratamiento de la arritmia
3.4.2.3. Disfunción del nódulo sinusal, bloqueo auriculoventricular y retraso de la conducción infrahisiana
3.4.2.4. Muerte súbita cardiaca y estratificación del riesgo
3.4.3. Hipertensión pulmonar
3.4.3.1. Introducción y clasificación
3.4.3.2. Diagnóstico
3.4.3.2.1. Estudio diagnóstico de la hipertensión pulmonar en las cardiopatías congénitas del adulto
3.4.3.2.2. Evaluación del riesgo
3.4.3.3. Tratamiento de la hipertensión pulmonar en las cardiopatías congénitas del adulto
3.4.3.3.1. Centros con experiencia
3.4.3.3.2. Medidas generales
3.4.3.3.3. Anticoagulación
3.4.3.3.4. Reparación del cortocircuito
3.4.3.3.5. Tratamiento médico para la hipertensión arterial pulmonar
3.4.4. Tratamiento quirúrgico
3.4.5. Cateterismo intervencionista
3.4.6. Endocarditis infecciosa
3.4.7. Tratamiento antitrombótico
3.4.8. Tratamiento de los pacientes cianóticos
3.4.8.1. Mecanismos adaptativos
3.4.8.2. Trastorno multisistémico
3.4.8.3. Presentación clínica e historia natural
3.4.8.4. Complicaciones tardías
3.4.8.5. Aspectos diagnósticos
3.4.8.6. Precauciones de laboratorio
3.4.8.7. Indicaciones para la intervención
3.4.8.8. Tratamiento médico
3.4.8.9. Recomendaciones sobre el seguimiento
3.4.8.10. Consideraciones adicionales
3.5. Consideraciones adicionales
3.5.1. Diferencias por sexo
3.5.2. Cardiopatías congénitas del adulto a edades avanzadas
3.5.3. Planificación anticipada de la atención y los cuidados paliativos
3.5.4. Seguro médico y empleo
3.5.5. Ejercicio y deporte
3.5.6. Cirugía no cardiaca
3.5.7. Embarazo, anticoncepción y asesoramiento genético
3.5.7.1. Embarazo y anticoncepción
3.5.7.2. Asesoramiento genético y riesgo de recurrencia
4. Lesiones específicas
4.1. Comunicación interauricular y conexión venosa pulmonar anómala
4.1.1. Introducción y antecedentes
4.1.2. Presentación clínica el historia natural
4.1.3. Estudio diagnóstico
4.1.4. Tratamiento quirúrgico/cateterismo intervencionista
4.1.5. Aspectos específicos de la conexión venosa pulmonar anómala aislada
4.1.6. Recomendaciones sobre el seguimiento
4.1.7. Consideraciones adicionales
4.2. Comunicación interventricular
4.2.1. Introducción y antecedentes
4.2.2. Presentación clínica e historia natural
4.2.3. Estudio diagnóstico
4.2.4. Tratamiento quirúrgico/cateterismo intervencionista
4.2.5. Recomendaciones sobre el seguimiento
4.2.6. Consideraciones adicionales
4.3. Defecto del septo auriculoventricular
4.3.1. Introducción y antecedentes
4.3.2. Presentación clínica e historia natural
4.3.3. Estudio diagnóstico
4.3.4. Tratamiento quirúrgico/cateterismo intervencionista
4.3.5. Recomendaciones sobre el seguimiento
4.3.6. Consideraciones adicionales
4.4. Ductus arterioso persistente
4.4.1. Introducción y antecedentes
4.4.2. Presentación clínica e historia natural
4.4.3. Estudio diagnóstico
4.4.4. Tratamiento quirúrgico/cateterismo intervencionista
4.4.5. Recomendaciones sobre el seguimiento
4.4.6. Consideraciones adicionales
4.5. Obstrucción del tracto de salida del ventrículo izquierdo
4.5.1. Estenosis valvular aórtica
4.5.1.1. Introducción y antecedentes
4.5.1.2. Presentación clínica e historia natural
4.5.1.3. Estudio diagnóstico
4.5.1.4. Tratamiento médico
4.5.1.5. Tratamiento quirúrgico/cateterismo intervencionista
4.5.1.6. Recomendaciones sobre el seguimiento
4.5.1.7. Consideraciones adicionales
4.5.2. Estenosis aórtica supravalvular
4.5.2.1. Introducción y antecedentes
4.5.2.2. Presentación clínica e historia natural
4.5.2.3. Estudio diagnóstico
4.5.2.4. Tratamiento quirúrgico/cateterismo intervencionista
4.5.2.5. Recomendaciones sobre el seguimiento
4.5.2.6. Consideraciones adicionales
4.5.3. Estenosis subaórtica
4.5.3.1. Introducción y antecedentes
4.5.3.2. Presentación clínica e historia natural
4.5.3.3. Estudio diagnóstico
4.5.3.4. Tratamiento quirúrgico/cateterismo intervencionista
4.5.3.5. Recomendaciones sobre el seguimiento
4.5.3.6. Consideraciones adicionales
4.6. Coartación aórtica
4.6.1. Introducción y antecedentes
4.6.2. Presentación clínica e historia natural
4.6.3. Estudio diagnóstico
4.6.4. Tratamiento quirúrgico/cateterismo intervencionista
4.6.5. Recomendaciones sobre el seguimiento
4.6.6. Consideraciones adicionales
4.7. Aortopatías
4.7.1. Síndrome de Marfan y otras enfermedades hereditarias de la aorta torácica
4.7.1.1. Introducción y antecedentes
4.7.1.2. Presentación clínica e historia natural
4.7.1.3. Estudio diagnóstico
4.7.1.4. Tratamiento médico
4.7.1.5. Tratamiento quirúrgico
4.7.1.6. Recomendaciones sobre el seguimiento
4.7.1.7. Consideraciones adicionales
4.7.2. Válvula aórtica bicúspide
4.7.3. Síndrome de Turner
4.8. Obstrucción del tracto de salida del ventrículo derecho
4.8.1. Introducción y antecedentes
4.8.2. Presentación clínica e historia natural
4.8.3. Estudio diagnóstico
4.8.4. Tratamiento quirúrgico/cateterismo intervencionista
4.8.5. Recomendaciones sobre el seguimiento
4.8.6. Consideraciones adicionales
4.9. Anomalía de Ebstein
4.9.1. Introducción y antecedentes
4.9.2. Presentación clínica e historia natural
4.9.3. Estudio diagnóstico
4.9.4. Tratamiento quirúrgico/cateterismo intervencionista
4.9.5. Recomendaciones sobre el seguimiento
4.9.6. Consideraciones adicionales
4.10. Tetralogía de Fallot
4.10.1. Introducción y antecedentes
4.10.2. Presentación clínica e historia natural
4.10.3. Estudio diagnóstico de los pacientes reparados
4.10.4. Tratamiento quirúrgico/cateterismo intervencionista tardíos
4.10.5. Indicaciones para estudio electrofisiológico e implante de desfibrilador
4.10.6. Recomendaciones sobre el seguimiento
4.10.7. Consideraciones adicionales
4.11. Atresia pulmonar con defecto del septo ventricular
4.11.1. Introducción y antecedentes
4.11.2. Presentación clínica e historia natural
4.11.3. Estudio diagnóstico
4.11.4. Tratamiento quirúrgico/cateterismo intervencionista
4.11.5. Recomendaciones sobre el seguimiento
4.11.6. Consideraciones adicionales
4.12. Transposición de las grandes arterias
4.12.1. Introducción y antecedentes
4.12.2. Operación de switch auricular
4.12.2.1. Presentación clínica tras el switch auricular
4.12.2.2. Estudio diagnóstico
4.12.2.3. Tratamiento médico
4.12.2.4. Tratamiento quirúrgico/cateterismo intervencionista
4.12.3. Operación de switch arterial
4.12.3.1. Presentación clínica tras el switch arterial
4.12.3.2. Estudio diagnóstico
4.12.3.3. Tratamiento quirúrgico/cateterismo intervencionista
4.12.4. Operación de tipo Rastelli
4.12.4.1. Presentación clínica tras la reparación de tipo Rastelli
4.12.4.2. Estudio diagnóstico
4.12.4.3. Tratamiento quirúrgico/cateterismo intervencionista
4.12.5. Recomendaciones sobre el seguimiento (independientemente del tipo de reparación)
4.12.6. Consideraciones adicionales (independientemente del tipo de reparación)
4.13. Transposición de las grandes arterias congénitamente corregida
4.13.1. Introducción y antecedentes
4.13.2. Presentación clínica e historia natural
4.13.3. Estudio diagnóstico
4.13.4. Tratamiento médico
4.13.5. Tratamiento quirúrgico/cateterismo intervencionista
4.13.6. Recomendaciones sobre el seguimiento
4.13.7. Consideraciones adicionales
4.14. Conducto del ventrículo derecho a la arteria pulmonar
4.14.1. Introducción y antecedentes
4.14.2. Estudio diagnóstico
4.14.3. Tratamiento quirúrgico/cateterismo intervencionista
4.14.4. Recomendaciones sobre el seguimiento
4.14.5. Consideraciones adicionales
4.15. Corazón univentricular
4.15.1. Introducción y antecedentes
4.15.2. Presentación clínica e historia natural
4.15.3. Estudio diagnóstico
4.15.4. Tratamiento conservador
4.15.5. Recomendaciones sobre el seguimiento
4.15.6. Consideraciones adicionales
4.16. Pacientes tras operación de Fontan
4.16.1. Introducción y antecedentes
4.16.2. Presentación clínica e historia natural
4.16.3. Estudio diagnóstico
4.16.4. Tratamiento médico
4.16.5. Tratamiento quirúrgico/cateterismo intervencionista
4.16.6. Recomendaciones sobre el seguimiento
4.16.7. Consideraciones adicionales
4.17. Anomalías coronarias
4.17.1. Introducción y antecedentes
4.17.1.1. Arteria coronaria anómala con origen en la arteria pulmonar
4.17.1.2. Origen aórtico anómalo de una arteria coronaria
4.17.1.3. Fístulas coronarias
4.17.2. Evaluación diagnóstica
4.17.3. Tratamiento quirúrgico
5. Indicadores de calidad
6. Lagunas en la evidencia
6.1. Aspectos generales
6.1.1. Planificación del cuidado y evaluación del paciente
6.1.2. Insuficiencia cardiaca
6.1.3. Arritmias
6.1.4. Hipertensión arterial pulmonar
6.1.5. Pacientes cianóticos
6.2. Complicaciones específicas
6.2.1. Cortocircuitos
6.2.2. Obstrucción del tracto de salida del ventrículo izquierdo y coartación
6.2.3. Aortopatías
6.2.4. Obstrucción del tracto de salida del ventrículo derecho
6.2.5. Anomalía de Ebstein
6.2.6. Tetralogía de Fallot
6.2.7. Transposición de las grandes arterias
6.2.8. Transposición de las grandes arterias congénitamente corregida
6.2.9. Corazón univentricular y operación de Fontan
6.2.10. Anomalías coronarias
7. Mensajes clave
7.1. Aspectos generales
7.1.1. Planificación de la atención y evaluación del paciente
7.1.2. Insuficiencia cardiaca
7.1.3. Arritmia
7.1.4. Hipertensión arterial pulmonar
7.1.5. Pacientes cianóticos
7.2. Complicaciones específicas
7.2.1. Cortocircuitos
7.2.2. Obstrucción del tracto de salida del ventrículo izquierdo
7.2.3. Coartación aórtica
7.2.4. Aortopatías
7.2.5. Obstrucción del tracto de salida del ventrículo derecho
7.2.6. Anomalía de Ebstein
7.2.7. Tetralogía de Fallot
7.2.8. Transposición de las grandes arterias
7.2.9. Transposición de las grandes arterias congénitamente corregida
7.2.10. Corazón univentricular y operación de Fontan
7.2.11. Anomalías coronarias
8. Mensajes clave de la guía sobre qué hacer y qué no
9. Apéndice
10. Bibliografía
Recomendaciones
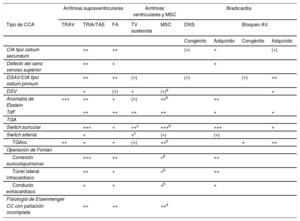
Recomendaciones para el tratamiento de las arritmias en las cardiopatías congénitas del adulto
Recomendaciones para el tratamiento de la hipertensión arterial pulmonar asociada con cardiopatía congénita
Recomendaciones para la intervención en la comunicación interauricular (congénita y residual)
Recomendaciones para la intervención en el defecto del septo ventricular (congénito o residual)
Recomendaciones para la intervención en el defecto del septo auriculo ventricular
Recomendaciones para la intervención en el ductus arterioso persistente
Recomendaciones para la intervención en la estenosis aórtica
Recomendaciones para la intervención en la estenosis aórtica supravalvular
Recomendaciones para la intervención en la estenosis subaórtica
Recomendaciones para la intervención en la coartación y recoartación aórtica
Recomendaciones para la cirugía aórtica en las aortopatías
Recomendaciones para la intervención en la obstrucción del tracto de salida del ventrículo derecho
Recomendaciones para la intervención en la anomalía de Ebstein
Recomendaciones para la reintervención tras reparación de la tetralogía de Fallot
Recomendaciones para la intervención en la transposición de las grandes arterias tras switch auricular
Recomendaciones para la intervención en la transposición de las grandes arterias tras switch arterial
Recomendaciones para la intervención en la transposición de las grandes arterias congénitamente corregida
Recomendaciones para la intervención en pacientes con conductos del ventrículo derecho a la arteria pulmonar
Consideraciones y recomendaciones especiales para la intervención en el corazón univentricular
Consideraciones y recomendaciones especiales para la intervención tras la operación de Fontan
Recomendaciones para el tratamiento de pacientes con arterias coronarias anómalas
Lista de tablas
Tabla 1 Clases de recomendaciones
Tabla 2 Niveles de evidencia
Tabla 3 Recomendaciones seleccionadas revisadas (R), recomendaciones nuevas (N) y conceptos nuevos
Tabla 4 Clasificación de la complejidad de las cardiopatías congénitas
Tabla 5 Requisitos de personal para los centros especializados en CCA
Tabla 6 Indicaciones de la resonancia magnética cardiovascular para pacientes con CCA
Tabla 7 Estimaciones del riesgo de eventos arrítmicos y bradicardias en las CCA
Tabla 8 Definiciones de los subtipos de hipertensión pulmonar y su aparición en las CCA
Tabla 9 Estrategias de reducción del riesgo de los pacientes con cardiopatía congénita cianótica
Tabla 10 Cardiopatía congénita con riesgo alto y extremadamente alto para el embarazo
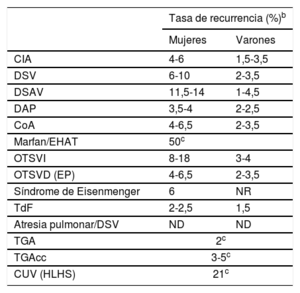
Tabla 11 Tasas de recurrencia de diversas lesiones cardiacas congénitas según el sexo de los padres afectadosTabla 12 Criterios diagnósticos del grado de estenosis aórtica
Lista de figuras
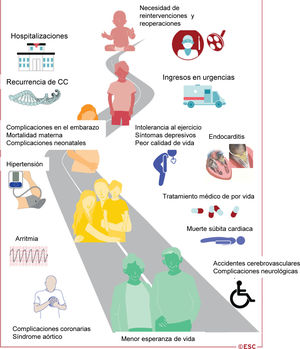
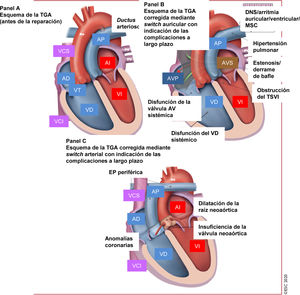
Figura 1 Ilustración central. La cardiopatía congénita es una enfermedad crónica que dura toda la vida
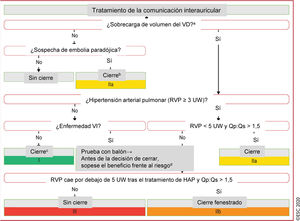
Figura 2 Tratamiento de la comunicación interauricular
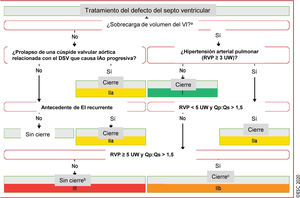
Figura 3 Tratamiento del defecto del septo ventricular.
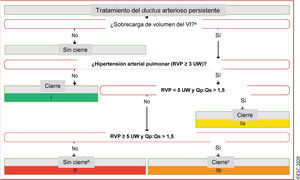
Figura 4 Tratamiento del ductus arterioso persistente
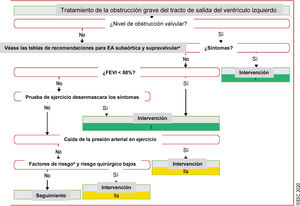
Figura 5 Tratamiento de la obstrucción grave del tracto de salida del ventrículo izquierdo.
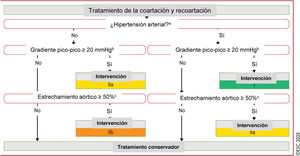
Figura 6 Tratamiento de la coartación y recoartación aórtica.
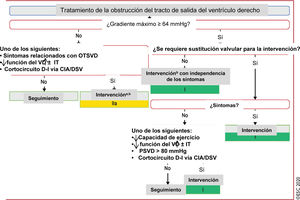
Figura 7 Tratamiento de la obstrucción del tracto de salida del ventrículo derecho
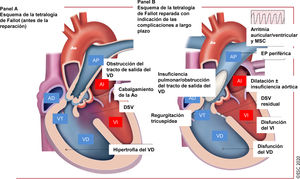
Figura 8 Tratamiento de la tetralogía de Fallot reparada: complicaciones a largo plazo que atender en el seguimiento.
Figura 9 Tratamiento de la transposición de las grandes arterias: complicaciones a largo plazo que atender en el seguimiento.
1PREÁMBULOLas guías tienen como objetivo reunir y evaluar toda la evidencia relevante disponible durante el proceso de elaboración sobre un tema determinado para ayudar a los médicos a seleccionar la mejor estrategia posible de tratamiento para un paciente en particular que sufre una enfermedad concreta. Las guías y las recomendaciones deben ayudar a los profesionales de la salud en la toma de decisiones clínicas en su ejercicio diario. No obstante, la decisión final sobre un paciente concreto debe tomarla el médico responsable de su salud, en consulta con el propio paciente o, cuando proceda, con la persona encargada de sus cuidados.
En los últimos años, la Sociedad Europea de Cardiología (ESC), además de otras sociedades y organizaciones científicas, ha publicado un gran número de guías. Debido al impacto de estas, se han establecido criterios de calidad para su elaboración de modo que todas las decisiones se presenten de manera clara y transparente al usuario. Las recomendaciones de la ESC para la elaboración y publicación de guías están disponibles en la sección de guías de la página web de la ESC (http://www.escardio.org/Guidelines-&-Education/Clinical-Practice-Guidelines/Guidelines-development/Writing-ESC-Guidelines). Las guías de la ESC representan la postura oficial de la ESC sobre un tema particular y se actualizan con regularidad.
Además de la publicación de las guías de práctica clínica, la ESC desarrolla el EurObservational Research Programme sobre registros internacionales de enfermedades e intervenciones cardiovasculares, que es esencial para evaluar los procesos diagnósticos/terapéuticos, el uso de los recursos y la adherencia a las guías. El objetivo de estos registros es mejorar la comprensión de la práctica médica en Europa y el mundo, con base en datos de alta calidad recogidos durante el ejercicio clínico diario.
Además, la ESC ha desarrollado e incluido en este documento una serie de indicadores de calidad, herramientas que permiten evaluar el grado de implementación de las guías. Estos indicadores pueden usarlos la ESC, hospitales, servicios públicos y profesionales de la salud para cuantificar la práctica clínica, así como en programas educativos junto con los mensajes clave de las guías para mejorar la calidad de la atención y los resultados clínicos.
Los miembros de este grupo de trabajo han sido seleccionados por la ESC y han incluido una representación de las subespecialidades más relevantes, para representar a los profesionales dedicados a los cuidados médicos de los pacientes con esta enfermedad. Los expertos seleccionados realizaron una revisión exhaustiva de la evidencia publicada sobre la atención a una entidad concreta según las normas establecidas por el comité de la ESC para la elaboración de las guías. Además, se llevó a cabo la evaluación crítica de los procedimientos diagnósticos y terapéuticos, incluida la valoración del cociente riesgo/beneficio. Se valoraron el nivel de evidencia y la fuerza de la recomendación de una opción terapéutica particular según escalas predefinidas, tal como se indica más adelante.
Los miembros del panel de redacción y los revisores del documento han declarado por escrito cualquier relación que se pueda considerar conflicto de intereses real o potencial. Estas declaraciones escritas están archivadas y disponibles en la página web de la ESC (http://www.escardio.org/guidelines). Este proceso garantiza la transparencia y evita posibles sesgos en los procesos de desarrollo y revisión. Durante el periodo de redacción, las modificaciones en las relaciones que se pudieran considerar conflicto de intereses se notificaron a la ESC y se actualizaron. El informe del grupo de trabajo fue financiado en su totalidad por la ESC y se desarrolló sin ninguna participación de la industria.
El comité para la elaboración de las guías de la ESC supervisa y coordina la preparación de nuevas guías. El comité es responsable también del proceso de aprobación de las guías. El comité de la ESC y expertos externos realizaron una revisión exhaustiva del documento, tras lo cual fue aprobado por todos los miembros del grupo de trabajo. Por último, el documento final fue aprobado por el comité de la ESC para su publicación en European Heart Journal. La elaboración de la presente guía se realizó tras la meticulosa evaluación del conocimiento científico y médico y de la evidencia disponible hasta la fecha de su redacción.
La tarea de elaboración de las guías incluye no solo la integración de la investigación más reciente, sino también la creación de herramientas educativas y programas de implementación de las recomendaciones. Para su implementación, se desarrollan ediciones de bolsillo, resúmenes en diapositivas y tarjetas, folletos con mensajes clave y versiones electrónicas para aplicaciones digitales (smartphones, etc.). Estas versiones son resumidas y, por lo tanto, en caso de necesidad debe consultarse la versión completa que se encuentra disponible gratuitamente en las páginas web de la ESC y European Heart Journal. Se recomienda a las sociedades nacionales que forman parte de la ESC suscribir, traducir e implementar las guías de la ESC. Los programas de implementación son necesarios porque se ha demostrado que los resultados clínicos se ven favorablemente influidos por la aplicación de las recomendaciones clínicas.
Se recomienda a los profesionales de la salud que tengan en consideración la presente guía de la ESC en la toma de decisiones clínicas en su ejercicio diario, así como en la determinación y la implementación de estrategias preventivas, diagnósticas y terapéuticas; no obstante, la decisión final sobre el cuidado de un paciente concreto, en consulta con dicho paciente y, si fuera necesario, con su representante legal, debe tomarla el médico responsable de su cuidado. Además, es responsabilidad del profesional de la salud comprobar la normativa aplicable a fármacos y dispositivos médicos antes de su prescripción.
2INTRODUCCIÓN2.1¿Por qué se necesita una guía nueva sobre el tratamiento de las cardiopatías congénitas del adulto?Desde la publicación de la versión anterior de la guía para el tratamiento de las cardiopatías congénitas del adulto (CCA) en 2010, se ha acumulado nueva evidencia para este grupo de pacientes, en particular sobre técnicas de intervención percutánea y estratificación del riesgo en relación con el momento de la cirugía y el cateterismo, y sobre el tratamiento médico, lo que ha hecho necesario actualizar las recomendaciones.
Hay un número creciente de pacientes en edad adulta e incluso ancianos con cardiopatías congénitas. A lo largo del documento se emplea el término CCA, tal como aparece en las publicaciones internacionales.
2.2Contenido de la guíaLa toma de decisiones en las CCA incluye el diagnóstico preciso, la planificación del momento óptimo para la intervención, la evaluación del riesgo y la estrategia de intervención más adecuada. Además, deben abordarse aspectos específicos del tratamiento médico para otras condiciones, como la insuficiencia cardiaca, la hipertensión pulmonar (HP) y la anticoagulación.
Esta guía se centra en las CCA y se orienta a su abordaje y tratamiento. Para más detalles sobre endocarditis, valvulopatía aislada y enfermedad aórtica, es preciso consultar las respectivas guías específicas publicadas por la ESC.
2.3Nuevo formato de la guíaLa guía nueva se ha adaptado para facilitar su uso en la práctica clínica y satisfacer las demandas de los profesionales centrándose en unas recomendaciones que se presentan de manera clara y condensada. Al final del documento, se proponen temas pendientes de investigación (apartado 5) y se resumen los mensajes clave (apartado 6). Para más información general, consulte el Libro de texto de Medicina Cardiovascular de la ESC1.
2.4Cómo usar esta guíaEl comité hace hincapié en el hecho de que muchos factores determinan en última instancia el tratamiento más apropiado para cada paciente individual dentro de una comunidad determinada. Estos factores incluyen la disponibilidad de equipos de diagnóstico, la experiencia de cardiólogos y cirujanos, especialmente en el campo de la cirugía cardiaca congénita y la intervención percutánea, y sobre todo los deseos de los pacientes bien informados. Además, debido a la falta de datos basados en la evidencia en el campo de las CCA, la mayoría de las recomendaciones son en gran medida el resultado del consenso de expertos basado en estudios y registros observacionales retrospectivos y prospectivos. Por lo tanto, en determinadas circunstancias clínicas puede ser adecuado desviarse de la presente guía. tablas 1–2
Clases de recomendaciones
| Definición | Expresiones propuestas | |
|---|---|---|
| Clase I | Evidencia y/o acuerdo general en que determinado procedimiento diagnóstico/tratamiento es beneficioso, útil y efectivo | Se recomienda/está indicado |
| Clase II | Evidencia conflictiva y/o divergencia de opinión acerca de la utilidad/eficacia de determinado tratamiento o procedimiento | |
| Clase IIa | El peso de la evidencia/opinión está a favor de su utilidad/eficacia | Se debe considerar |
| Clase IIb | La utilidad/eficacia está menos establecida por la evidencia/opinión | Se puede considerar |
| Clase III | Evidencia o acuerdo general en que determinado tratamiento no es útil/efectivo y en algunos casos puede ser perjudicial | No se recomienda |
Niveles de evidencia
| Nivel de evidencia A | Datos procedentes de múltiples ensayos clínicos aleatorizados o metanálisis |
|---|---|
| Nivel de evidencia B | Datos procedentes de un único ensayo clínico aleatorizado o de grandes estudios no aleatorizados |
| Nivel de evidencia C | Consenso de opinión de expertos y/o pequeños estudios, estudios retrospectivos, registros |
En la tabla 3 se presenta un resumen de las recomendaciones revisadas seleccionadas, las recomendaciones nuevas y los conceptos nuevos.
Recomendaciones seleccionadas revisadas (R), recomendaciones nuevas (N) y conceptos nuevos
| Arritmias | ||
|---|---|---|
| N | No había recomendaciones formales sobre arritmias en la guía de 2010 (ahora se han incluido). Para conocer los detalles, véase las tablas de los apartados 3.4.2 y 4.10. Este es el resumen de los aspectos más importantes: | |
| • Se enfatiza la importancia de comprender la causa, el mecanismo de la arritmia y la anatomía de la CC subyacente | ||
| • Se enfatiza la importancia del enfoque multidisciplinario para el tratamiento óptimo de la arritmia previo o simultáneo a las intervenciones percutáneas o quirúrgicas | ||
| • Se considera la ablación precoz con catéter como alternativa al tratamiento médico a largo plazo para la TSV y la TV sintomáticas siempre que el procedimiento se realice en centros con experiencia | ||
| • Se propone la ablación de los istmos anatómicos relacionados con la TV en pacientes con TdF reparada y TV sostenida antes de la reintervención percutánea o quirúrgica de la OTSVD, ya que estos procedimientos pueden dificultar/inhabilitar el acceso a los sustratos de la TV | ||
| • Se reconoce la asociación entre bradicardia y TRIA y el beneficio potencial de implantar un MP | ||
| Síndrome de Eisenmenger/hipertensión arterial pulmonar | ||
| N | Se recomienda desaconsejar el embarazo a las pacientes con CCA e HP precapilar confirmada | |
| N | Está recomendada la evaluación del riesgo de todo paciente con CCA-HAP | |
| N | Para los pacientes con riesgo bajo e intermedio con lesiones simples reparadas e HAP, se recomienda tratamiento oral combinado inicial o tratamiento combinado secuencial; los pacientes con riesgo alto deben recibir tratamiento combinado inicial que incluya prostanoides parenterales | |
| R | Se enfatiza la importancia del tratamiento secuencial de la HAP en el síndrome de Eisenmenger y la PM6M para la decisión de iniciar el tratamiento | Para los pacientes con síndrome de Eisenmenger y capacidad de esfuerzo reducida (distancia en la PM6M < 450 m), se debe considerar una estrategia basada en monoterapia inicial con antagonistas del receptor de la endotelina seguida de tratamiento combinado si el paciente no mejora |
| Cortocircuitos | ||
| N | Es obligatoria la determinación invasiva de la RVP de los pacientes con cortocircuitos y signos no invasivos de elevación de la PAP | |
| N/R | Las recomendaciones ajustadas para el cierre del cortocircuito (cuando Qp:Qs > 1,5) según la RVP calculada son: | |
| • < 3 UW: clase I para CIA, DSV y DAP | ||
| • 3-5 UW: clase IIa para CIA, DSV y DAP | ||
| • ≥ 5 UW pero disminuyendo a < 5 UW después del tratamiento de la HAP: clase IIb para CIA (solo cierre fenestrado) | ||
| • ≥ 5 UW para DSV y DAP (decisión individualizada y cuidadosa en centros con experiencia): clase IIb | ||
| • ≥ 5 UW a pesar del tratamiento de la HAP: clase III para CIA | ||
| N | Para los pacientes con CIA y enfermedad del VI, se recomienda hacer prueba con balón y sopesar cuidadosamente el beneficio de eliminar el cortocircuito I-D con el impacto potencialmente negativo del cierre de la CIA debido al aumento de la presión de llenado (considerando la posibilidad de cierre, cierre fenestrado o ausencia de cierre) | |
| N | Se debe tener en cuenta la edad más avanzada en la decisión del cierre quirúrgico de la CIA | Para pacientes ancianos no aptos para cierre con dispositivo, se recomienda sopesar el riesgo quirúrgico con el beneficio potencial del cierre de la CIA |
| R | El cierre percutáneo del DSV se ha convertido en una alternativa al cierre quirúrgico en pacientes seleccionados, sobre todo en el DSV residual | El cierre percutáneo del DSV se ha convertido en una alternativa, sobre todo en el DSV residual, el DSV con mal acceso quirúrgico y el DSV muscular localizado en el centro del septo interventricular |
| R | Especificar la necesidad de que sea un cirujano especialista en CC quien realice el cierre parcial del DSAV | El cierre quirúrgico está recomendado para pacientes con importante sobrecarga de volumen del VD y solo puede realizarlo un cirujano especialista en CC |
| R | Especificar que la FA o la HP son requisitos para la reparación valvular en el DSAV | Para pacientes asintomáticos con importante insuficiencia de la válvula AV del lado izquierdo, función del VI conservada (DTSVI < 45 mm o FEVI > 60%), probabilidad alta de reparación valvular exitosa y riesgo quirúrgico bajo, se debe considerar la intervención en presencia de FA o PAP sistólica > 50 mmHg |
| R | Especificar la opción de cierre fenestrado de la CIA | En pacientes con RVP ≥ 5 UW, se puede considerar el cierre fenestrado de la CIA cuando la RVP caiga por debajo de 5 UW después del tratamiento de la HAP y haya cortocircuito I-D relevante (Qp:Qs > 1,5) |
| R | Incluir la desaturación durante el ejercicio como contraindicación para el cierre de la CIA, DSV, DSAV y DAP | No se recomienda el cierre del cortocircuito en pacientes con HAP grave (RVP ≥ 5 UW) que presentan desaturación durante el ejercicio |
| Obstrucción del tracto de salida del ventrículo izquierdo y aortopatías | ||
| R | Aumentar la clase de recomendación para la intervención de la EA de bajo flujo y bajo gradiente desde IIa a I | Está indicada la intervención para pacientes con EA grave de bajo flujo y bajo gradiente (gradiente medio < 40 mmHg) y FE reducida con evidencia de reserva (contráctil) de flujo que excluye la EA seudograve |
| R | Reducir el umbral del gradiente Doppler medio para la intervención de la OTSVI de 50 a 40 mmHg | Está recomendada la cirugía para pacientes sintomáticos con EA valvular, subvalvular o supravalvular y un gradiente Doppler medio ≥ 40 mmHg |
| R | Incluir la concentración de BNP y el aumento de la PAP en la indicación para la intervención de la EA valvular | Se debe considerar la intervención de los pacientes asintomáticos con FE normal sin anomalías en la prueba de ejercicio (véase el apartado 4.5.1) cuando el riesgo quirúrgico sea bajo y alguno de los siguientes hallazgos esté presente: |
| • Concentración de BNP notablemente elevada (> 3 veces el valor normal corregido por sexo y edad), confirmado por determinaciones repetidas y sin otra explicación | ||
| • HP grave (PAP sistólica en reposo > 60 mmHg confirmada por determinación invasiva) sin otra explicación | ||
| R | Confirmar los gradientes de presión por determinaciones invasivas; se prefiere el implante de stent cuando sea técnicamente factible en la coartación y la recoartación | Está indicada la reparación de la coartación o la recoartación (quirúrgica o percutánea) de pacientes hipertensos con aumento del gradiente (no invasivo) entre las extremidades superiores e inferiores confirmado por una determinación invasiva (pico-pico ≥ 20 mmHg), con preferencia por el tratamiento percutáneo (stent) cuando sea técnicamente factible |
| N | Se debe considerar un tratamiento percutáneo (stent) para la coartación de pacientes normotensos con aumento del gradiente confirmado por una determinación invasiva (pico-pico ≥ 20 mmHg) cuando sea técnicamente factible | |
| N | En las aortopatías, está recomendada la reparación de la válvula aórtica mediante reimplante o remodelado con anuloplastia aórtica para pacientes jóvenes con síndrome de Marfan o EHAT relacionada con la dilatación de la raíz aórtica y la reparación con válvulas aórticas tricuspídeas cuando la realicen cirujanos experimentados | |
| N | Se debe considerar la cirugía para los pacientes con mutación TGFBR1 o TGFBR2 (incluyendo el síndrome de Loeys-Dietz) que tienen enfermedad de la raíz aórtica con un diámetro máximo del seno aórtico ≥ 45 mm | |
| N | En el síndrome de Turner, se debe considerar la cirugía electiva para los aneurismas de la raíz aórtica o la aorta ascendente de mujeres mayores de 16 años con un ITA ascendente > 25 mm/m2 y factores de riesgo de disección aórtica | |
| N | En el síndrome de Turner, se puede considerar la cirugía electiva para los aneurismas de la raíz aórtica o la aorta ascendente de mujeres mayores de 16 años con un ITA ascendente > 5 mm/m2 que no tengan factores de riesgo de disección aórtica | |
| Obstrucción del tracto de salida del ventrículo derecho/tetralogía de Fallot/Ebstein | ||
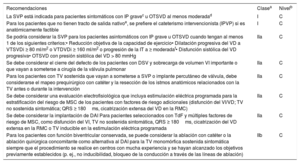
| R | Ajustar las recomendaciones para la intervención quirúrgica en la OTSVD según los síntomas | Si la sustitución valvular quirúrgica es la única opción, está indicada para pacientes con estenosis grave que estén sintomáticos |
| Si la sustitución valvular quirúrgica es la única opción para pacientes con estenosis grave que están asintomáticos, está indicada cuando se cumplen uno o más de los siguientes requisitos: | ||
| • Reducción objetiva de la capacidad de ejercicio | ||
| • Función del VD decreciente o RT progresiva hasta moderada como mínimo | ||
| • Presión sistólica del VD > 80 mmHg | ||
| • Cortocircuito D-I por CIA o DSV | ||
| R | Incluir preferencia por el cateterismo intervencionista para implante de válvula pulmonar en la TdF | Para los pacientes sin tracto de salida nativo, se prefiere la IPVP cuando sea anatómicamente factible |
| R | Especificar la dilatación del VD en el contexto de la sustitución de la válvula pulmonar para la TdF y los conductos del TSVD | Se debe considerar la sustitución de la válvula pulmonar de los pacientes asintomáticos con RP grave u OTSVD, en presencia de dilatación progresiva del VD a VTSVDi ≥ 80 ml/m2, VTDVDi ≥ 160 ml/m2 o progresión de la RT hasta moderada como mínimo |
| R | En caso de CIA en la anomalía de Ebstein, aumente la precaución sobre el incremento de la presión de la AD o la caída del gasto cardiaco | En caso de embolia sistémica documentada probablemente causada por embolia paradójica, se debe considerar el cierre aislado con dispositivo de la CIA/FOP, aunque se requiere una evaluación cuidadosa antes de la intervención para excluir la inducción del aumento de presión de la AD o la caída del gasto cardiaco |
| Cuando la cianosis (saturación de oxígeno en reposo < 90%) sea el problema dominante, se puede considerar solo el cierre con dispositvo de la CIA/FOP, aunque se requiere una evaluación cuidadosa antes de la intervención para excluir la inducción de un aumento de presión de la AD o la caída del gasto cardiaco | ||
| Transposición de las grandes arterias | ||
| R | Reducir el nivel de recomendación para la reparación de la válvula AV en TGA/switch auricular de I a IIa en pacientes sintomáticos | Para los pacientes con insuficiencia de la válvula AV sistémica grave (tricuspídea) sin disfunción sistólica ventricular relevante (FE > 40%), se debe considerar la reparación o sustitución valvular independientemente de los síntomas |
| R | En pacientes con TGA/switch auricular que requieren implante de MP/DAI, conviene prestar especial atención a posibles fugas del conducto | Para pacientes con una fuga del conducto que requieren un MP/DAI, se debe considerar el cierre de la fuga cuando sea técnicamente posible y antes de la inserción de los cables transvenosos |
| N | En la TGAcc, se debe considerar la estimulación biventricular en caso de bloqueo AV completo o si se requiere estimulación ventricular > 40% | |
| R | Revisar las indicaciones de sustitución de VT en la TGAcc, según síntomas y función ventricular sistémica (actualizar el nivel de recomendación para pacientes con TGAcc sintomático de IIa a I) | Para los pacientes sintomáticos con IT grave y función sistólica del VD sistémico conservada o levemente alterada (FE > 40%), está indicado el reemplazo de la VT |
| Para los pacientes asintomáticos con IT grave y dilatación progresiva del VD sistémico o función sistólica del VD sistémico levemente alterada (FE > 40%), se debe considerar la sustitución de la VT | ||
| Para los pacientes sintomáticos con IT grave y función sistólica del VD sistémico más que levemente reducida (FE ≤ 40%), se puede considerar la sustitución de la VT | ||
| R | La reparación anatómica (switch auricular + arterial) para la TGAcc se ha eliminado de las recomendaciones | |
| Corazón univentricular | ||
| N | Se recomienda que los adultos con CUV no operados o paliados se sometan a una evaluación cuidadosa en centros especializados que incluya técnicas multimodales de imagen y estudios invasivos para decidir si pueden beneficiarse de procedimientos quirúrgicos o intervencionistas | |
| Circulación de Fontan | ||
| N | La arritmia auricular sostenida con conducción AV rápida es una urgencia médica y debe tratarse de inmediato con cardioversión eléctrica | |
| N | La anticoagulación está indicada en caso de trombo auricular, arritmias auriculares o eventos tromboembólicos, o antecedentes de estas condiciones | |
| N | Se debe desaconsejar el embarazo a las mujeres con circulación de Fontan y cualquier complicación | |
| N | Se recomienda realizar cateterismo cardiaco en un umbral bajo en casos de edema inexplicable, deterioro de la capacidad de ejercicio, arritmia de nueva aparición, cianosis y hemoptisis | |
| N | Para los pacientes con arritmias, se debe considerar un enfoque proactivo de evaluación electrofisiológica y ablación (cuando sea apropiado) | |
| N | Se deben considerar las pruebas periódicas de imagen hepática (ecografía, tomografía computarizada, resonancia magnética) | |
| N | Se puede considerar los antagonistas del receptor de la endotelina y los inhibidores de la fosfodiesterasa-5 para pacientes seleccionados con presión o resistencia pulmonar elevadas en ausencia de aumento de la presión ventricular telediastólica | |
| N | Para pacientes seleccionados con cianosis significativa, se puede considerar el cierre con dispositivo de una fenestración, pero se requiere una evaluación cuidadosa antes de la intervención para excluir la inducción de un aumento de la presión venosa sistémica o una caída del gasto cardiaco | |
| Anomalías coronarias | ||
| N | Se recomienda obtener imágenes funcionales no farmacológicas (p. ej., estudio nuclear, ecocardiografía o RMC con estrés físico) de los pacientes con anomalías coronarias para confirmar/excluir la isquemia miocárdica | |
| A. Coronaria anómala con origen en la arteria pulmonar (CAAP) | ||
| N | Se recomienda la cirugía para los pacientes con ALCAPA | |
| N | Se recomienda la cirugía para los pacientes con ARCAPA y síntomas atribuibles a la coronaria anómala | |
| N | Se debe considerar la cirugía de la ARCAPA para los pacientes asintomáticos con disfunción ventricular o isquemia miocárdica atribuible a una anomalía coronaria | |
| B. Origen aórtico anómalo de una arteria coronaria (OAAAC) | ||
| N | Se recomienda la cirugía de OAAAC para los pacientes con síntomas típicos de angina que muestren evidencia de isquemia miocárdica inducida por estrés en un territorio coincidente o anatomía de alto riesgo | |
| N | Se debe considerar la cirugía para los pacientes asintomáticos con OAAAC (derecha o izquierda) y evidencia de isquemia miocárdica | |
| N | Se debe considerar la cirugía para los pacientes asintomáticos con OAAACI sin evidencia de isquemia miocárdica pero con una anatomía de alto riesgo | |
| N | Se puede considerar la cirugía para los pacientes sintomáticos con OAAAC incluso cuando no haya evidencia de isquemia miocárdica ni anatomía de alto riesgo | |
| N | Se puede considerar la cirugía para los pacientes asintomáticos con OAAACI sin isquemia miocárdica y sin anatomía de alto riesgo cuando se presentan a una edad temprana (< 35 años) | |
| N | No está recomendada la cirugía de OAAACD en pacientes asintomáticos sin isquemia miocárdica ni anatomía de alto riesgo | |
| Conceptos nuevos | ||
| Nomenclatura (CCA) | ||
| Clasificación de la complejidad de las enfermedades | ||
| Requisitos de personal para un centro especializado en CCA | ||
| Papel emergente de los biomarcadores en el seguimiento de las CCA | ||
| Recomendaciones detalladas y específicas para el tratamiento de las arritmias | ||
| Recomendaciones más específicas y ajustadas para el tratamiento de la HAP | ||
| Recomendaciones sobre el uso de anticoagulantes | ||
| Consideraciones sobre el envejecimiento y la planificación anticipada de la atención | ||
| Categorías de embarazos de alto riesgo según la guía sobre embarazo | ||
| Se expande el apartado del síndrome de Marfan a las aortopatías (y se incluyen la EHAT, el síndrome de Turner y la enfermedad aórtica bicúspide) | ||
| Papel emergente del cateterismo intervencionista en las CCA | ||
AD: auricular/auricular derecha; ALCAPA: coronaria izquierda anómala desde la arteria pulmonar; ARCAPA: coronaria derecha anómala desde la arteria pulmonar; AV: auriculoventricular; BNP: péptido natriurético cerebral; CAAP: coronaria anómala con origen en la arteria pulmonar; CC: cardiopatía congénita; CCA: cardiopatía congénita del adulto; CIA: comunicación interauricular; CUV: corazón univentricular; DAI: desfibrilador automático implantable; DAP: ductus arterioso persistente; D-I: derecha-izquierda; DSAV: defecto del septo auriculoventricular; DSV: defecto del septo ventricular; DTSVI: diámetro telesistólico del ventrículo izquierdo; EA: estenosis aórtica; EHAT: enfermedad hereditaria de la aorta torácica; FA: fibrilación auricular; FE: fracción de eyección; FEVI: fracción de eyección del ventrículo izquierdo; FOP: foramen oval permeable; HAP: hipertensión arterial pulmonar; HAP-CC: hipertensión arterial pulmonar asociada con cardiopatía congénita; HP: hipertensión pulmonar; I–D: izquierda-derecha; IPVP: implante percutáneo de la válvula pulmonar; IT: insuficiencia tricuspídea; ITA: índice de tamaño aórtico; N: recomendación nueva; OAAAC: origen aórtico anómalo de una arteria coronaria; OAAACI: origen aórtico anómalo de la coronaria izquierda; OTSVD: obstrucción del tracto de salida del ventrículo derecho; OTSVI: obstrucción del tracto de salida del ventrículo izquierdo; PAP: presión arterial pulmonar; PM6M: prueba de los 6 min de marcha; Qp:Qs: cociente de flujo pulmonar a sistémico; R: recomendación revisada; RMC: resonancia magnética cardiaca; RP: regurgitación pulmonar; RVP: resistencia vascular pulmonar; TdeF: tetralogía de Fallot; TGA: transposición de las grandes arterias; TGAcc: transposición de las grandes arterias congénitamente corregida; TGFBR: receptor del factor de crecimiento transformador beta; TRIA: taquicardia por reentrada intraauricular; TSV: taquicardia supraventricular; TSVD: tracto de salida del ventrículo derecho; TSVI: tracto de salida del ventrículo izquierdo; TV: taquicardia ventricular; VD: ventrículo derecho; VI: ventrículo izquierdo; VT: válvula tricúspide; VTDVDi: volumen telediastólico del ventrículo derecho indexado; VTSVDi: volumen telesistólico del ventrículo derecho indexado.
Actualmente la prevalencia mundial de las cardiopatías congénitas (CC) es de alrededor de 9/1.000 nacimientos, con una importante variabilidad geográfica2,3. Si bien la prevalencia de las malformaciones cardiacas congénitas graves está disminuyendo en muchos países occidentales/desarrollados gracias a la detección precoz intrauterina y la interrupción del embarazo, la prevalencia general a escala mundial está aumentando4. Debido al desarrollo médico, quirúrgico y tecnológico de las últimas décadas, más del 90% de las personas nacidas con CC sobreviven hasta la edad adulta5. Como consecuencia, la prevalencia de las CC en la población ha aumentado y actualmente supera con creces la tasa de mortalidad infantil6. Las CC se clasifican en leves, moderadas o graves (tabla 4).
Clasificación de la complejidad de las cardiopatías congénitas
| Leve |
| • Enfermedad valvular aórtica congénita aislada y enfermedad de la válvula aórtica bicúspide |
| • Enfermedad congénita aislada de la válvula mitral (excepto válvula en paracaídas, orificio en forma de hendidura) |
| • Estenosis pulmonar aislada leve (infundibular, valvular, supravalvular) |
| • CIA, DSV o DAP pequeño y aislado |
| • CIA de tipo ostium secundum reparada, defecto del seno venoso, DSV o DAP sin secuelas, como agrandamiento de la cámara, disfunción ventricular o PAP elevada |
| Moderada (reparada o no reparada cuando no se especifique, en orden alfabético): |
| • Conexión venosa pulmonar anómala (parcial o total) |
| • Coronaria anómala con origen en la AP |
| • Coronaria anómala con origen en el seno contrario |
| • Estenosis aórtica-subvalvular o supravalvular |
| • DSAV, parcial o completo, incluida la CIA tipo ostium primum (excluida la enfermedad vascular pulmonar) |
| • CIA tipo ostium secundum moderada o grande no reparada (excluida la enfermedad vascular pulmonar) |
| • Coartación de la aorta |
| • Ventrículo derecho de doble cámara |
| • Anomalía de Ebstein |
| • Síndrome de Marfan y EHAT relacionadas, síndrome de Turner |
| • DAP moderado o grande no reparado (excluida la enfermedad vascular pulmonar) |
| • Estenosis pulmonar periférica |
| • Estenosis pulmonar (infundibular, valvular, supravalvular) moderada o grave |
| • Aneurisma/fístula del seno de Valsalva |
| • Defecto del seno venoso |
| • Tetralogía de Fallot reparada |
| • Transposición de las grandes arterias tras operación de switch arterial |
| • DSV con anomalías asociadas (excluida la enfermedad vascular pulmonar) o cortocircuito moderado o grave |
| Grave (reparada o no reparada cuando no se especifique, en orden alfabético) |
| • Cualquier CC (reparada o no reparada) asociada con enfermedad vascular pulmonar (incluyendo síndrome de Eisenmenger) |
| • Cualquier CC cianótica (no operada o paliada) |
| • Ventrículo de doble salida |
| • Circulación de Fontan |
| • Arco aórtico interrumpido |
| • Atresia pulmonar (todas las formas) |
| • Transposición de las grandes arterias (excepto pacientes con operación de switch arterial) |
| • Corazón univentricular (ventrículo izquierdo/derecho de doble entrada, atresia tricuspídea/mitral, síndrome del corazón izquierdo hipoplásico o cualquier otra anomalía anatómica con un único ventrículo funcional) |
| • Tronco arterioso |
| • Otras anomalías complejas de la conexión AV y ventriculoarterial (como el corazón entrecruzado, síndromes de heterotaxia o inversión ventricular) |
AP: arteria pulmonar; AV: auriculoventricular; CC: cardiopatía congénita; CIA: comunicación interauricular; DAP: ductus arterioso persistente; DSAV: defecto del septo AV; DSV: defecto del septo ventricular; EHAT: enfermedad hereditaria de la aorta torácica; PAP: presión arterial pulmonar; VI: ventrículo izquierdo.
Cuando los pacientes con CC se acercan a la edad adulta, deben ser transferidos a la unidad de atención de CCA. Esta transferencia debe ir precedida de una fase de transición preparatoria, que continúa hasta la edad adulta según las necesidades del paciente. Se requieren programas de formación y organización sanitaria específicos para satisfacer las necesidades de esta población de pacientes7. Es importante señalar que la atención de los pacientes con CCA es un proceso que dura toda la vida (figura 1) y requiere estrategias de planificación de la atención por adelantado. El grupo de trabajo de Cardiopatías Congénitas en el Adulto de la ESC ha publicado un documento de posición de expertos con recomendaciones para la organización de la atención y la formación en la subespecialidad de CCA en Europa8. Este documento se refiere a la guía previa de la ESC9 y estratifica la atención de los pacientes en 3 niveles: a) pacientes que requieren atención exclusivamente en centros especializados; b) pacientes para los que se puede establecer una atención compartida con los servicios cardiacos generales para adultos, y c) pacientes a los que se puede tratar en centros no especializados (con acceso a atención especializada si es necesario). Los requisitos propuestos para el personal de los centros especializados se describen en la tabla 5. La complejidad de la malformación cardiaca no debería ser el único criterio para asignar a los pacientes un determinado nivel de atención. Aunque a los pacientes con malformaciones complejas se les puede asignar fácilmente un nivel de atención alto, ciertos defectos anatómicamente simples pueden requerir atención especializada en determinadas circunstancias (p. ej., el defecto del septo auricular con hipertensión arterial pulmonar [HAP]). Por lo tanto, se recomienda que todos los pacientes con CCA sean atendidos una vez en un centro especializado, lo que permite a los especialistas en CCA determinar el nivel de atención más adecuado y los intervalos de seguimiento en cada caso8. Deben establecerse redes de centros especializados con atención general para adultos en cada área de influencia. De hecho, con el aumento del número de adultos con CC, va a haber cada vez más pacientes que acudan por primera vez a los cardiólogos generales por afecciones agudas, como arritmia, insuficiencia cardiaca o endocarditis. En tales casos, los cardiólogos generales no deben retrasar el tratamiento de los pacientes hemodinámicamente inestables, y deben ponerse en contacto inmediatamente con el centro de CCA del paciente para establecer las estrategias de tratamiento más adecuadas o el traslado del paciente. Es necesaria una atención especial para los pacientes que se han sometido a la corrección de Fontan y presentan arritmias, ya que incluso las arritmias supraventriculares no se toleran bien. Próximamente van a publicarse las recomendaciones detalladas sobre la atención de urgencia para pacientes con CCA en un documento de posición de expertos independiente.
 MSC: muerte súbita cardiaca.' title='Ilustración central. La cardiopatía congénita es una enfermedad crónica que dura toda la vida. CC: cardiopatía congénita;
MSC: muerte súbita cardiaca.' title='Ilustración central. La cardiopatía congénita es una enfermedad crónica que dura toda la vida. CC: cardiopatía congénita; Requisitos de personal para los centros especializados en CCA*
| Disciplina | Número requerido |
|---|---|
| Cardiólogo adulto/pediátrico con certificación en CCA | ≥ 2 |
| Especialista en imagen de CCA (certificación en ETT/ETE, RMC, TCC) | ≥ 2 |
| Cardiólogo intervencionista especializado en CC | ≥ 2 |
| Cirujano especializado en CC | ≥ 2 |
| Anestesiólogo con experiencia y conocimientos en CC | ≥ 2 |
| Enfermera especializada (si los organismos nacionales de enfermería profesional permiten la especialización) | ≥ 2 |
| Electrofisiólogo invasivo con experiencia en CCA | ≥ 1 |
| Experto en enfermedad vascular pulmonar | ≥ 1 |
| Genetista clínico | ≥ 1 |
| Psicólogo | ≥ 1 |
| Trabajador social | ≥ 1 |
| Equipo de cuidados paliativos |
CC: cardiopatía congénita; CCA: cardiopatías congénitas del adulto; ETE: ecocardiografía transesofágica; ETT: ecocardiografía transtorácica; RMC: resonancia magnética cardiovascular; TCC: tomografía computarizada cardiovascular.
*Modificado de Baumgartner et al.8.
La transferencia de adolescentes a la unidad de CCA es vital a medida que se acercan a la edad adulta sin que se produzcan interrupciones en la atención, y debe ir precedida de una fase de transición preparatoria con un apoyo adicional que se prolongue hasta la edad adulta temprana según las necesidades del paciente10.
La transición requiere una organización sanitaria especial10,11. Se recomienda que los centros especializados en CCA tengan equipos que incluyan enfermeras especializadas, psicólogos y trabajadores sociales, ya que la ansiedad, la depresión o los problemas para afrontar la enfermedad son preocupaciones comunes de los pacientes adultos con CC12. Estos profesionales también desempeñan un papel fundamental en el proceso de transición al hacerse cargo de la atención de los pacientes después de su transferencia desde la cardiología pediátrica. Los aspectos que deben abordar los profesionales sanitarios aliados incluyen la salud mental, el bienestar psíquico y la calidad de vida12,13. A lo largo del proceso de atención de las CCA, las opciones y estrategias de planificación relacionadas con los cuidados paliativos también requieren el apoyo de expertos.
3.3Estudio diagnósticoPara el estudio diagnóstico de los pacientes con CCA, además del examen clínico completo, es muy importante revisar la historia clínica, incluida la información detallada sobre la cirugía paliativa o reparadora y el cateterismo intervencionista. El objetivo de analizar la historia del paciente es evaluar los síntomas presentes y pasados, así como buscar episodios intercurrentes y cualquier cambio en la medicación. Los síntomas más frecuentes de los pacientes con CCA son la intolerancia al esfuerzo y las palpitaciones. La capacidad física autopercibida no se corresponde adecuadamente con la cuantificación objetiva de la capacidad de esfuerzo14. Por este motivo, las pruebas de ejercicio cardiopulmonar (PECP) han ganado protagonismo en la evaluación objetiva de la intolerancia al esfuerzo de pacientes tanto aparentemente asintomáticos como sintomáticos. Se debe preguntar al paciente por sus hábitos de vida para detectar cambios progresivos en la actividad diaria y limitar así la subjetividad del análisis de los síntomas. En pacientes sintomáticos, se debe tener en cuenta (y excluir cuando sea necesario) otras causas, como anemia, depresión, ganancia de peso y desentrenamiento físico, además de malformaciones congénitas y sus secuelas.
El examen clínico tiene un papel muy importante e incluye una cuidadosa evaluación de cualquier cambio en los hallazgos auscultatorios y la presión arterial o el desarrollo de signos de insuficiencia cardiaca. Tanto el electrocardiograma (ECG) como la oximetría de pulso se realizan habitualmente junto con el examen clínico. La radiografía torácica proporciona información sobre los cambios en el tamaño y la configuración del corazón y la vascularización pulmonar. Las técnicas de imagen no invasivas más habituales son la ecocardiografía transtorácia (ETT) con ecocardiografía transesofágica (ETE) y resonancia magnética cardiovascular (RMC) cuando esté indicado. Los pacientes con CCA pueden beneficiarse especialmente de los marcapasos y desfibriladores compatibles con resonancia magnética.
La ecocardiografía es superior a la RMC para valorar los gradientes de presión y la presión arterial pulmonar (PAP), y para detectar estructuras pequeñas y muy móviles como las vegetaciones. La RMC es ideal para la cuantificación precisa de los volúmenes ventriculares, la fracción de eyección (FE), la insuficiencia valvular15, el cálculo del flujo sanguíneo pulmonar y sistémico y la evaluación de la fibrosis miocárdica. La tomografía computarizada cardiovascular (CCT) con escáneres modernos de fuente única o doble se puede realizar con protocolos de ahorro de dosis y puede ser necesaria para indicaciones especiales, como se indica en la tabla 6. Es importante que haya una colaboración interdisciplinaria de expertos: los expertos en imagen de CC deben intercambiar opiniones con los cirujanos de CC, los intervencionistas y los electrofisiólogos para optimizar la contribución de las imágenes a la atención clínica, y deben trabajar conjuntamente en pos de un uso apropiado de las imágenes multimodales. Las imágenes avanzadas suelen reservarse para cuando los pacientes se atienden en el centro especializado, antes que repetirlas.
Indicaciones de la resonancia magnética cardiovascular para pacientes con CCA
| Indicaciones |
| • Quantificación de los volúmenes del VD, la FE (incluidos el VD subpulmonar, el VD sistémico y el ventrículo único) |
| • Evaluación de la OTSVD y los conductos VD-AP |
| • Cuantificación de la IP |
| • Evaluación de las AP (estenosis, aneurismas) y de la aorta (aneurisma, disección, coartación —la TCC puede ser superior—) |
| • Evaluación de las venas sistémicas y pulmonares (conexión anómala, obstrucción, anatomía venosa coronaria preprocedimiento, etc.) |
| • Colaterales y malformaciones arteriovenosas (la TCC puede ser superior) |
| • Anomalías coronarias y EC (la TCC es superior para el recorrido intramural, el recorrido en forma de hendidura, el despegue en ángulo agudo, el puente miocárdico y la evaluación de la placa) |
| • Detección y cuantificación de la isquemia miocárdica por RMC de perfusión de estrés |
| • Evaluación de masas intracardiacas y extracardiacas |
| • Cuantificación de la masa miocárdica (VI y VD) |
| • Detección y cuantificación de la fibrosis/cicatriz miocárdica (realce tardío de gadolinio, mapeo en T1), caracterización tisular (fibrosis, grasa, hierro, etc.) |
| • Cuantificación del flujo sanguíneo sistémico y pulmonar para calcular el Qp:Qs |
| • Cuantificación de la distribución de la perfusion a los pulmones derecho e izquierdo |
| • Determinación del flujo pulmonar en pacientes con múltiples fuentes de suministro sanguíneo (colaterales aortopulmonares principales) |
AP: arteria pulmonar; CC: cardiopatía congénita; CCA: cardiopatías congénitas del adulto; EC: enfermedad coronaria; IP: insuficiencia pulmonar; OTSVD: obstrucción del tracto de salida del ventrículo derecho; Qp:Qs: cociente de flujo pulmonar a sistémico; RMC: resonancia magnética cardiovascular; TCC: tomografía computarizada cardiovascular; VD: ventrículo derecho; VI: ventrículo izquierdo.
La ecocardiografía, la RMC y la TCC requieren personal con experiencia en CC y en imagen, lo que implica formación y recursos. Dentro de la ESC, esta formación está reconocida por un examen de certificación de la European Association of Cardiovascular Imaging (EACVI), independiente del examen estándar de la ETT, ETE o RMC, y específico para las CC.
3.3.1EcocardiografíaLa ecocardiografía sigue siendo el examen de primera línea16. Las modalidades de ecocardiografía en modo M, ecocardiografía bidimensional y ecocardiografía tridimensional (3D) se usan para imagen, mientras que la imagen de Doppler tisular y sus derivados, especialmente deformación longitudinal y velocidad de deformación, se han convertido en parte integral de la evaluación funcional17.
La ecocardiografía ofrece información sobre la anatomía cardiaca básica, incluidas la orientación y la posición del corazón, el retorno venoso, la conexión de aurículas y ventrículos y el origen de las grandes arterias. Para la evaluación de la morfología y la función de las válvulas cardiacas, la ETT y, cuando sea necesario, la ETE (actualmente combinada con ecocardiografía 3D) es la modalidad de imagen de elección. Esto también se aplica a los cortocircuitos, como en el caso de la comunicación interauricular (CIA) o los defectos del septo ventricular (DSV); la ecocardiografía 3D permite tener una perspectiva frontal, lo que facilita la evaluación del tamaño y la forma de la malformación y su relación con las estructuras circundantes.
La ETT permite medir y calcular el tamaño y el volumen ventriculares y la FE. También es capaz de detectar la sobrecarga de volumen cuando existe cortocircuito o insuficiencia valvular, así como la sobrecarga de presión en caso de aumento de la poscarga, con una buena calidad de imagen. Incluso las técnicas más antiguas que utilizan el modo M para medir el desplazamiento sistólico del plano del anillo tricuspídeo y el desplazamiento sistólico del plano del anillo mitral siguen siendo válidas, especialmente durante el seguimiento. Para la función sistólica del ventrículo izquierdo (VI), la ecocardiografía 3D, la imagen de Doppler tisular y la imagen de deformación bidimensional se han demostrado como instrumentos robustos y merecen integrarse a la práctica clínica. Incluso teniendo en cuenta las técnicas más nuevas, la ecocardiografía sigue siendo fundamental en la evaluación del seguimiento de la función sistólica de un ventrículo derecho (VD) o único, aunque para obtener mediciones más precisas, a menudo se necesitan imágenes adicionales obtenidas por RMC.
3.3.2Resonancia magnética cardiovascularLa RMC se ha convertido en una técnica esencial en las unidades especializadas. Permite una reconstrucción anatómica 3D excelente, que no se ve restringida por el tamaño corporal o las ventanas acústicas, con una resolución espacial y temporal en continua mejora18. Si bien una calidad de imagen óptima requiere un ritmo cardiaco regular, los estudios diagnósticos con RMC suelen realizarse en pacientes con ritmo cardiaco irregular (ectopia frecuente o fibrilación auricular [FA]) y artefactos metálicos. La RMC es el método de imagen estándar por excelencia para la cuantificación de los volúmenes. Puede ser una alternativa cuando no es posible obtener imágenes ecocardiográficas de suficiente calidad o como método complementario cuando las mediciones ecocardiográficas resultan dudosas o ambiguas. Al no producir radiación, es útil en casos de evaluaciones seriadas (p. ej., para la monitorización de las dimensiones aórticas). La RMC permite calcular el flujo sanguíneo sistémico y pulmonar de pacientes con múltiples fuentes de suministro sanguíneo y, cuando se combina con procedimientos de cateterismo invasivo, permite determinar la resistencia vascular pulmonar (RVP). La RMC es especialmente útil para caracterizar la fibrosis miocárdica. Cada vez se emplea mmás la RMC con realce tardío de gadolinio para la fibrosis focal y las imágenes de mapeo T1 para la fibrosis intersticial de pacientes con CCA, por su potencial diagnóstico y su valor pronóstico. Se están llevando a cabo grandes estudios de CC sobre malformaciones específicas para determinar si esta tecnología es capaz de predecir la supervivencia.
Se debe evitar el realce de gadolinio en pacientes con una tasa de filtrado glomerular baja (< 30ml/min/1,73 m2) para minimizar el riesgo de fibrosis sistémica nefrogénica. Por ello, se recomienda determinar la concentración de creatinina antes de realizar la RMC. Aunque todavía no se han observado repercusiones clínicas, la posibilidad de que se produzcan depósitos cerebrales de gadolinio a largo plazo (independientemente de la función renal) ha despertado preocupación sobre las dosis acumuladas a lo largo de la vida en pacientes con CC que se someten a RMC seriadas desde edades tempranas. Teniendo en cuenta estos factores, es preferible que el gadolinio se administre selectivamente en centros especializados y con un contraste macrocíclico, en lugar de un agente lineal, ya que se ha observado menor retención de este metal con aquel19.
Los adultos con CC portadores de marcapasos (MP) y desfibriladores (DAI) convencionales pueden someterse a RMC siguiendo las recomendaciones donde esté disponible apoyo local20.
La imagen 3D por RMC se puede integrar en los procedimientos electrofisiológicos para guiarlos. Las reconstrucciones 3D por TCC y RMC también pueden ser útiles para los ensayos de realidad virtual o para planificar las intervenciones a partir de impresión 3D en pacientes específicos.
Las indicaciones para la RMC se resumen en la tabla 6.
3.3.3Tomografía computarizada cardiovascularLa TCC ofrece una resolución espacial excelente y un tiempo de obtención corto. Es especialmente relevante para la obtención de imágenes de grandes arterias, coronarias y arterias colaterales, así como en la enfermedad del parénquima pulmonar (tabla 6). En muchas instituciones, la TCC es la modalidad de imagen preferida para planificar el implante percutáneo de valvulas. Permite evaluar el tamaño y la función ventriculares con menos resolución temporal que la RMC, aunque con una gran dosis de radiación, por lo que no debe practicarse en serie en este contexto. Los avances recientes han reducido sustancialmente la cantidad de exposición a la radiación, hasta < 5 mSv para una combinación de TCC coronaria, pulmonar y angiograma aórtico. Esta posibilidad ha convertido la TCC en una opción más atractiva en indicaciones específicas para pacientes con CCA, sobre todo enfermedad coronaria, y la evaluación detallada de las colaterales21.
La TCC es particularmente útil en una situación urgente como la disección aórtica, la embolia pulmonar y el absceso paravalvular en la endocarditis, en los que puede tener ventajas respecto a la ecocardiografía y la RMC por ser menos susceptible a los artefactos de las válvulas protésicas.
En los pacientes con válvulas protésicas (in situ > 3 meses), la tomografía por emisión de positrones con fluorodesoxiglucosa/TC es útil para el diagnóstico precoz de inflamación e infección en el lugar de la válvula y para identificar focos infecciosos en otras localizaciones22.
3.3.4Pruebas de esfuerzo cardiopulmonarLa prueba formal de esfuerzo es importante en la población con CCA, cuyas calidad de vida y capacidad funcional son indicadores clave del éxito de la intervención. La prueba de ejercicio cardiopulmonar (PECP), que incluye la evaluación de la capacidad objetiva de ejercicio (captación máxima de oxígeno), la eficacia de la ventilación (pendiente VE/VCO2), la respuesta cronotrópica y la presión arterial, así como de la arritmia inducida por el ejercicio, ofrece una valoración más amplia de la función y el estado físico y tiene objetivos principales que se correlacionan bien con la morbididad y la mortalidad de los pacientes con CCA23. Las pruebas de esfuerzo seriadas, por lo tanto, deben formar parte de los protocolos de seguimiento a largo plazo y los ensayos de intervención. Son importantes para planificar las intervenciones y las reintervenciones. La PECP también es útil para establecer las recomendaciones personalizadas sobre la intensidad del ejercicio físico24. La prueba de los 6min de marcha (PM6M) es otra prueba sencilla que permite cuantificar la capacidad de esfuerzo físico; se relaciona con los resultados clínicos en pacientes con HAP.
3.3.5Cateterismo cardiacoActualmente el cateterismo cardiaco se reserva para la resolución de cuestiones anatómicas y fisiológicas específicas o para las intervenciones. Las indicaciones incluyen la evaluación de la RVP, la función diastólica ventricular (incluyendo la fisiología constrictiva y restrictiva), los gradientes de presión y la cuantificación del cortocircuito cuando la evaluación no invasiva resulta dudosa, la angiografía coronaria y la evaluación de vasos extracardiacos como las arterias colaterales aortopulmonares.
En los cortocircuitos con hipertensión pulmonar documentada por ecocardiografía Doppler, el cateterismo con prueba de vasorreactividad sigue siendo esencial para la toma de decisiones terapéuticas. El óxido nítrico inhalado es el agente de elección en estos casos. La estimación de la RVP en los cortocircuitos requiere un cálculo preciso del flujo pulmonar mediante el principio de Fick. Este método, con el cálculo del consumo de oxígeno, es el que permite la cuantificación más precisa del gasto cardiaco.
Antes de la cirugía, se debe tomar imágenes de la arteria coronaria (por TCC o angiografía) de los varones mayores de 40 años, mujeres posmenopáusicas y todo paciente con al menos 1 factor de riesgo de enfermedad coronaria (EC)25.
3.3.6BiomarcadoresEn los pacientes con CC hay diferentes clases de biomarcadores que se asocian con eventos adversos, como las neurohormonas y los marcadores de daño miocárdico (troponinas de alta sensibilidad) o inflamación (proteína C reactiva de alta sensibilidad). Entre las neurohormonas, los péptidos natriuréticos (péptido natriurético cerebral [BNP] o el fragmento aminoterminal del propéptido natriurético cerebral [NT-pro-BNP]) son los más estudiados en pacientes con CCA. Tienen un valor pronóstico importante, pero son menos útiles para diagnosticar insuficiencia cardiaca en las diferentes malformaciones cardiacas debido a que presentan una variabilidad que depende del defecto subyacente y el tipo de reparación26. Son más útiles en pacientes con circulación biventricular y menos útiles en pacientes con circulación de Fontan27. Las determinaciones seriadas de los péptidos natriuréticos ayudan a identificar a los pacientes con riesgo de eventos adversos. Es importante tener en cuenta que los péptidos natriuréticos pueden estar aumentados en pacientes cianóticos debido a que la hipoxia induce su secreción28.
3.4Consideraciones terapéuticas3.4.1Insuficiencia cardiacaLa insuficiencia cardiaca es una complicación frecuente que afecta al 20-50% de los pacientes con CCA, y es su principal causa de muerte29. Su incidencia está aumentando y probablemente esté subestimada. Dado que es frecuente que haya signos y síntomas latentes de insuficiencia cardiaca, se debe hacer un seguimiento y un cribado sistemático de los pacientes con riesgo alto de insuficiencia cardiaca30. Es necesario excluir y, si es posible, tratar mediante intervencionismo o cirugía cualquier anomalía hemodinámica que pueda ser causa de insuficiencia cardiaca, como las arritmias. En general, los médicos de CCA intentan seguir las recomendaciones vigentes para el tratamiento de la insuficiencia cardiaca y las comorbilidades relacionadas, como la diabetes mellitus, la FA, la apnea del sueño, la deficiencia de hierro y la caquexia31. No obstante, puesto que la fisiopatología de la disfunción cardiorrespiratoria suele ser muy diferente de la circulación «normal» defectuosa, extrapolar los resultados de los estudios publicados a los pacientes con CCA puede ser difícil, sobre todo aquellos con VD sistémico, ventrículo subpulmonar defectuoso o fisiología de ventrículo único. La fisiopatología de la insuficiencia cardiaca en las CCA con disfunción ventricular sistólica incluye un amplio espectro de causas. Tanto los ventrículos sistémicos como los subpulmonares, ya sean izquierdos o derechos, incluidos los ventrículos únicos, pueden sufrir sobrecarga crónica de presión o volumen, lo que conduce a una disfunción ventricular progresiva. La arquitectura miocárdica alterada (sin compactación) y la interdependencia ventricular pueden comprometer la función ventricular sistólica. Se puede producir daño miocárdico (menor protección miocárdica durante la cirugía de revascularización, tras una ventriculotomía y después de una hipoxia crónica) en pacientes con CCA. Por último, la cardiopatía isquémica (relacionada principalmente con el envejecimiento o la presencia de malformaciones coronarias congénitas) y la taquiarritmia persistente pueden ser causa de una alteración en la función ventricular sistémica y subpulmonar30. Los pocos datos disponibles sobre el tratamiento para la insuficiencia cardiaca de pacientes con CCA no son concluyentes y proceden de pequeños grupos de pacientes. Por lo tanto, las recomendaciones específicas de la CCA suelen basarse en la experiencia clínica y los posicionamientos de expertos30. Cuando existe circulación biventricular, se suele dar a los pacientes con VI defectuoso sistémico el tratamiento convencional para la insuficiencia cardiaca; esto también es válido para los pacientes sintomáticos con VD defectuoso sistémico. Los diuréticos sirven más que nada para controlar los síntomas. Siguen sin establecerse los beneficios clínicos a largo plazo del tratamiento con inhibidores del sistema renina-angiotensina-aldosterona y los bloqueadores beta. Tampoco se conocen los beneficios clínicos a largo plazo del tratamiento convencional de la insuficiencia cardiaca en el ventrículo subpulmonar defectuoso, aunque los diuréticos pueden aliviar los síntomas. El tratamiento de los pacientes sintomáticos con un ventrículo único defectuoso en circulación de Fontan o cuando existe un cortocircuito derecha-izquierda (D-I) persistente debe iniciarse con precaución, teniendo en cuenta el equilibrio precario entre la precarga ventricular y la poscarga sistémica. Actualmente, solo unos pocos estudios de pequeño tamaño han investigado los beneficios del sacubitrilo-valsartán en pacientes con CCA e insuficiencia cardiaca. Se ha demostrado que este tratamiento reduce la morbimortalidad de la insuficiencia cardiaca crónica, por lo que se ha incluido recientemente en las recomendaciones de la guía de la ESC31. La insuficiencia cardiaca con FE conservada también es frecuente en los pacientes con CCA. El tratamiento en este contexto debe basarse en las recomendaciones terapéuticas generales para la insuficiencia cardiaca. La terapia de resincronización cardiaca (TRC) ha adquirido cada vez más interés para los pacientes con CCA e insuficiencia cardiaca congestiva. Hasta el momento hay pocas pruebas en que basar la definición de indicaciones y resultados. La eficacia de la TRC en las CC puede variar dependiendo del sustrato estructural y funcional, como la anatomía del ventrículo sistémico (izquierdo, derecho o único), la presencia y el grado de insuficiencia valvular auriculoventricular (AV) estructural sistémica, la enfermedad miocárdica primaria o cicatrización y el tipo de retraso de la conducción eléctrica32.
Se espera que la incidencia de la insuficiencia cardiaca aguda en pacientes con CCA también aumente con el tiempo debido al aumento de la edad de los pacientes y la presencia de enfermedades más complejas. El conocimiento del uso adecuado de inotrópicos, la disponibilidad de oxigenación extracorpórea y las técnicas avanzadas de bypass son requisitos indispensables para el soporte vital de los pacientes con CCA e insuficiencia cardiaca aguda. En estos casos, se recomienda la derivación a un centro especializado33.
Se puede considerar el trasplante cardiaco en casos de insuficiencia cardiaca terminal. Los resultados clínicos tras la cirugía de trasplante mejoran continuamente, sobre todo en pacientes con CCA, pero la mortalidad perioperatoria sigue siendo más alta que en otras enfermedades, sobre todo por las complicaciones derivadas de la cirugía cardiaca previa, la existencia de una fisiopatología y una anatomía complejas y la presencia de comorbilidades (enfermedad multisistémica). El uso creciente de dispositivos de asistencia ventricular puede servir como tratamiento puente hasta el trasplante. En algunos casos concretos, estos dispositivos pueden ser una opción permanente. Algunos pacientes pueden tener una anatomía extremadamente compleja o altas concentraciones de anticuerpos contra los antígenos leucocitarios humanos, por lo que no son candidatos para el trasplante.
Algunos pacientes pueden requerir un trasplante multiorgánico. El trasplante cardiopulmonar se aplica a pacientes con CC que tengan hipertensión arterial pulmonar (HAP) irreversible, como el síndrome de Eisenmenger; no obstante, la falta de donantes de órganos es una limitación importante. Raramente se realiza un trasplante simultáneo de corazón e hígado cuando se produce insuficiencia hepática tras la operación de Fontan o en la hepatopatía congestiva de larga duración por insuficiencia cardiaca derecha (p. ej. anomalía de Ebstein). Hay poca experiencia en este tipo de cirugía. En todos los casos, se recomienda que especialistas en insuficiencia cardiaca por CCA hagan una evaluación adecuada de la necesidad de trasplante en un centro de trasplantes con experiencia en CCA. Se debe ofrecer una planificación de la atención que incluya cuidados paliativos a todos los pacientes con insuficiencia cardiaca avanzada.
3.4.2Arritmias y muerte súbita cardiaca3.4.2.1Sustratos arritmogénicosEn los pacientes con CCA se puede encontrar todo el espectro de arritmias. No obstante, existen algunos sustratos arritmogénicos congénitos que tienen que ver con la propia malformación (véase la tabla 7 y el apartado 4). El aumento de la esperanza de vida (junto con la exposición a los habituales factores de riesgo de los sustratos arritmogénicos) incrementa la prevalencia de arritmias relacionadas con el remodelado estructural, que pueden ocurrir a una edad más temprana que en la población general, como la FA.
Estimaciones del riesgo de eventos arrítmicos y bradicardias en las CCA
| Arritmias supraventriculares | Arritmias ventriculares y MSC | Bradicardia | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Tipo de CCA | TRAV | TRIA/TAE | FA | TV sostenida | MSC | DNS | Bloqueo AV | ||
| Congénito | Adquirido | Congénito | Adquirido | ||||||
| CIA tipo ostium secundum | ++ | ++ | (+) | + | (+) | ||||
| Defecto del seno venoso superior | ++ | + | + | ||||||
| DSAV/CIA tipo ostium primum | ++ | ++ | (+) | (+) | (+) | ++ | |||
| DSV | + | (+) | + | (+)a | + | ||||
| Anomalía de Ebstein | +++ | ++ | + | (+) | ++b | ++ | |||
| TdF | ++ | ++ | ++ | ++ | + | + | |||
| TGA | |||||||||
| Switch auricular | +++ | + | ++c | +++b | +++ | + | |||
| Switch arterial | + | +c | (+) | (+) | |||||
| TGAcc | ++ | + | + | (+) | ++b | + | ++ | ||
| Operación de Fontan | |||||||||
| Conexión auriculopulmonar | +++ | ++ | +b | ++ | |||||
| Túnel lateral intracardiaco | ++ | + | +b | ++ | |||||
| Conducto extracardiaco | + | + | +b | + | |||||
| Fisiología de Eisenmenger | |||||||||
| CC con paliación incompleta | ++ | ++ | ++d | ||||||
(+): riesgo mínimo; +: riesgo leve; ++: riesgo moderado; +++: riesgo alto; AV: auriculoventricular; CC: cardiopatía congénita; CIA: comunicación interauricular; DNS: disfunción del nódulo sinusal; DSAV: defecto del septo AV; DSV: defecto del septo ventricular; FA: fibrilación auricular; MSC: muerte súbita cardiaca; TAE: taquicardia auricular ectópica; TdF: tetralogía de Fallot; TGA: transposición de las grandes arterias; TGAcc: transposición de las grandes arterias congénitamente corregida; TRAV: taquicardia por reentrada AV; TRIA: taquicardia por reentrada intraauricular; TV: taquicardia ventricular.
aTeniendo en cuenta la alta prevalencia de DSV, el riesgo global de los pacientes no seleccionados con DSV se considera mínimo.
bLa MSC puede deberse a arritmias supraventriculares con conducción AV rápida.
cRiesgo estimado de TV más alto en la dextro-TGA compleja.
dNo arrítmica.
Las celdas vacías indican que, aunque no se especifica, se producen eventos arrítmicos (no hay símbolo).
Otras arritmias se relacionan con el tipo y el momento de la reparación de la CCA. Las incisiones en la aurícula derecha (AD), junto con el remodelado secundario a la sobrecarga hemodinámica, contribuyen al aumento de la prevalencia de las taquicardias auriculares (TA) en distintas CC. La más frecuente es la taquicardia por reentrada intraauricular (TRIA) tardía, especialmente el flutter auricular dependiente del istmo cavotricuspídeo. Las velocidades auriculares de 150-250 lpm pueden producir una conducción AV rápida, deterioro hemodinámico y muerte súbita cardiaca (MSC). La taquicardia ventricular (TV) monomórfica también depende de la malformación (véase la tabla 7) y el tipo de reparación34,35, con la parte crítica del circuito de macrorreentrada típicamente ubicada dentro de istmos anatómicamente bien definidos y bordeados por cicatrices quirúrgicas y material de parche. Por el contrario, en los pacientes con insuficiencia progresiva del ventrículo sistémico o subpulmonar pueden aparecer cambios electrofisiológicos (EF) más complejos, que incluyan remodelado de canales iónicos, alteraciones en el calcio y cambios de la matriz extracelular que favorezcan la aparición de diferentes tipos de arritmias, como la TV polimórfica rápida y la fibrilación ventricular (FV)36.
3.4.2.2Evaluación de pacientes con arritmia sospechada o documentada y tratamiento de la arritmiaLa evaluación de los pacientes sintomáticos sin arritmia documentada en el momento de la visita depende de la frecuencia de los síntomas (registro Holter, consulta del dispositivo cuando lo haya, registro de episodios) y las circunstancias en que se han producido (prueba de esfuerzo).
No está clara la utilidad de la evaluación periódica más allá de los ECG de 12 derivaciones (como el Holter periódico) para los pacientes asintomáticos. Se ha documentado una gran prevalencia de hallazgos asintomáticos que raramente influyen en el tratamiento37.
Se debe evaluar en todos los pacientes si la causa de la arritmia es reversible (p. ej., hipertiroidismo o proceso inflamatorio) o hay anomalías hemodinámicas nuevas o residuales. Se debe terminar inmediatamente las arritmias que producen inestabilidad hemodinámica, independientemente de la duración o la anticoagulación, de acuerdo con las recomendaciones actuales32.
En algunos casos puede producirse parada/bradicardia sinusal posconversión, por lo que es necesario considerar la disponibilidad de dispositivos de estimulación de apoyo para los pacientes con riesgo de disfunción del nódulo sinusal (véase la tabla 7). Si se tolera la TRIA/FA y dura 48 h o más, hay que descartar un posible trombo cardiaco o iniciar una anticoagulación adecuada (> 3 semanas) con control farmacológico del ritmo antes de la cardioversión mediante un bloqueador beta o un antagonista de los canales de calcio (en pacientes con función ventricular sistémica normal que no tienen preexcitación)32,37,38. El objetivo terapéutico para todos los pacientes con CC es el mantenimiento del ritmo sinusal32,37. La ablación por catéter es el tratamiento de primera línea, por delante de los tratamientos farmacológicos crónicos, en caso de sustratos susceptibles y circunscritos, ya que los fármacos antiarrítmicos suelen asociarse con efectos inotrópicos o dromotrópicos negativos32. Los fármacos antiarrítmicos de clase 1C pueden reducir el ritmo de la TRIA sin bloquear la conducción AV, lo que permite una conducción 1:1 con empeoramiento hemodinámico32. Se puede considerar la amiodarona para la prevención de la recurrencia de TA/FA en pacientes con CC y disfunción ventricular sistémica, hipertrofia del ventrículo sistémico o EC, en los que la ablación no funciona o no se puede practicar. La amiodarona suele tener efectos secundarios y se debe usar con precaución en pacientes con cianosis, peso corporal bajo, enfermedad hepática, tiroidea o pulmonar o QT largo. No se recomienda el tratamiento prolongado con amiodarona para pacientes jóvenes con CC32.
Para el tratamiento óptimo de las arritmias crónicas, es obligatoria la derivación a un centro que disponga de un equipo multidisciplinario y experto en arritmias relacionadas con CC32,37. Véase el apartado 3.4.7. para obtener información más específica sobre la anticoagulación.
3.4.2.3Disfunción del nódulo sinusal, bloqueo auriculoventricular y retraso de la conducción infrahisianaSe debe considerar el implante periódico de Holter en pacientes asintomáticos con riesgo de disfunción del nódulo sinusal y bloqueo AV. La disfunción crónica del nódulo sinusal/bradicardia con hemodinámica auricular ineficaz puede producir remodelado auricular y TRIA. Los pacientes con bloqueo AV posoperatorio tienen mayor riesgo de MSC. En consecuencia, se han propuesto indicaciones más amplias para el implante de marcapasos en comparación con los pacientes que tienen corazones estructuralmente normales20,32.
Las indicaciones de TRC para pacientes con CCA y circulación biventricular y VI sistémico siguen los criterios estándar. Es importante señalar que la principal causa de disfunción ventricular sistémica es la estimulación ventricular convencional y no el bloqueo de rama. Por consiguiente, se recomienda la TRC para pacientes con CCA que tengan FE sistémica ≤ 35% y QRS estrecho, y se debe hacer previsión anticipada para la colocación de un dispositivo nuevo. También se puede considerar la estimulación hisiana como tratamiento alternativo. La eficacia de la TRC en las CCA varía según el tipo de anomalía y depende de la anatomía individual y de las causas de la disfunción (como VD sistémico/ventrículo único, insuficiencia de la válvula AV, cicatrización). En general, la duración del intervalo QRS por sí solo puede no ser un factor predictivo suficiente. Además, suelen ser necesarios una toracotomía o un implante híbrido y no se dispone de datos sobre duración32.
3.4.2.4Muerte súbita cardiaca y estratificación del riesgoLa MSC relacionada con arritmias ventriculares es un importante motivo de preocupación, ya que representa un 7-26% de todas las muertes de adultos29,39,40. Aunque la incidencia poblacional de las CC es relativamente baja (< 0,1% anual), algunas malformaciones colocan al paciente en una situación de riesgo alto (véase la tabla 7). La identificación de los pacientes con riesgo de MSC sigue siendo un reto.
El implante de un DAI para la prevención secundaria de la MS y la prevención primaria en pacientes con fisiología biventricular y VI sistémico sigue los criterios estándar37,41. Se puede emplear fármacos antiarrítmicos como tratamiento coadyuvante al DAI para reducir la carga arrítmica del ventrículo32. El beneficio del DAI para la prevención primaria en pacientes con VD sistémico o ventrículo único no se conoce bien.
En consecuencia, salvo la tetralogía de Fallot (TdF), siguen sin establecerse unas directrices específicas sobre implante de DAI para la prevención primaria en pacientes con CCA32,37. Aunque se han utilizado sistemas de DAI transvenosos, los subcutáneos son más adecuados para los pacientes con un acceso venoso al ventrículo difícil o un cortocircuito intracardiaco. Sin embargo, no todos los pacientes son candidatos, debido al riesgo de que la detección sea inadecuada y no se produzca una estimulación antitaquicárdica y antibradicárdica.
La utilidad de la estimulación eléctrica programada (EEP) en los pacientes con CC asintomáticos todavía es incierta. Parece adecuada cuando existen incisiones ventriculares o hay un sustrato para la reentrada ventricular que se encuentra típicamente, aunque no solo, en los pacientes con TdF reparada (TdFr). Es importante reconocer otras posibles causas de MSC por bradicardia/bloqueo AV total y arritmia ventricular inducida por bradicardia con o sin QT largo y TRIA/FA con conducción rápida.
Recomendaciones para el tratamiento de las arritmias en las cardiopatías congénitas del adulto
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Para los pacientes con CC de complejidad moderada y grave (tabla 4) y arritmias documentadas, está indicada la derivación a un centro que cuente con un equipo multidisciplinario especializado en CCA y en arritmias relacionadas con CCA | I | C |
| Para los pacientes con CC y arritmias documentadas o con alto riesgo de arritmias tras el procedimiento (p. ej., cierre de la CIA a una edad más avanzada) considerados para (re)intervenciones percutáneas o quirúrgicas, está indicada la derivación a un centro que cuente con un equipo multidisciplinario con experiencia en estas intervenciones y en el tratamiento invasivo de las arritmias | I | C |
| En la CC leve, se recomienda la ablación con catéter en lugar del tratamiento médico de larga duración para la TSV recurrente sostenida y sintomática (TRNAV, TRAV, TA y TRIA) o que podría implicar MSC (tabla 7) | I | C |
| En las CCA moderadas y graves, se debe considerar la ablación con catéter de la TSV recurrente sostenida y sintomática (TRNAV, TRAV, TA y TRIA) o que podría implicar MSC (tabla 7), siempre que el procedimiento se realice en centros con experiencia | IIa | C |
| La ablación con catéter está indicada como tratamiento complementario a los DAI para los pacientes con TV monomórfica recurrente, TV incesante o tormenta eléctrica no tratable con medicamentos o reprogramación del DAI | I | C |
| Se debe considerar la ablación con catéter de la TV sostenida monomórfica sintomática en pacientes no aptos para tratamiento médico, siempre que el procedimiento se realice en centros con experiencia | IIa | C |
| Desfibrilador automático implantable | ||
| Después de evaluar la causa del episodio y excluir las causas reversibles, está indicado el implante de un DAI para los adultos con CC que han sobrevivido a parada cardiaca por FV o TV no tolerada hemodinámicamente | I | C |
| Tras evaluación hemodinámica y reparación cuando corresponda, está indicado el implante de DAI para los adultos con CC y TV sostenida. El estudio EF es necesario para identificar a los pacientes para quienes la ablación percutánea o quirúrgica pueda ser beneficiosa como tratamiento complementario o una alternativa razonable | I | C |
| Se debe considerar el implante de DAI para los adultos con CC que tengan fisiología biventricular, VI sistémico e insuficiencia cardiaca sintomática (NYHA II/III) y FE ≤ 35% a pesar de al menos 3 meses de un tratamiento médico óptimo, siempre que tengan una expectativa de vida muy superior a 1 año en buen estado funcionalc | IIa | C |
| Se debe considerar el implante de DAI para los pacientes con CC y síncope de causa desconocida con sospecha de arritmia que tengan disfunción ventricular avanzada o TV/FV inducible en estimulación eléctrica programada | IIa | C |
| Se debe considerar el implante de DAI para los pacientes seleccionados con TdF y múltiples factores de riesgo de MSC, como disfunción del VI, TV sintomática no sostenida, QRS ≥ 180ms, cicatrización extensa del VD en la RMC o TV inducible en estimulación eléctrica programada | IIa | C |
| Se puede considerar el implante de DAI para los pacientes con disfunción avanzada del VD único o sistémico (FE sistémica del VD < 35%) en presencia de factores de riesgo adicionalesd | IIb | C |
| Marcapasos | ||
| Se debe considerar el implante de MP para los pacientes con CCA y síndrome de bradicardia-taquicardia para prevenir la TRIA si fracasa la ablación o no es posible llevarla a cabo | IIa | C |
| Se debe considerar el implante de MP para los pacientes con CCA grave y bradicardia sinusal o de unión (frecuencia cardiaca diurna < 40 lpm o pausas > 3 s) | IIa | C |
| Se debe considerar el implante de MP para los pacientes con CCA y deterioro hemodinámico debido a bradicardia sinusal o pérdida de la sincronía AV | IIa | C |
| Se puede considerar el implante de MP para pacientes con CCA moderada y bradicardia sinusal o de unión (frecuencia cardiaca diurna < 40 lpm o pausas > 3 s) | IIb | C |
AV: auriculoventricular; CC: cardiopatía congénita; CCA: cardiopatías congénitas del adulto; CIA: comunicación interauricular; DAI: desfibrilador automático implantable; EF: electrofisiológico; FE: fracción de eyección; FV: fibrilación ventricular; MP: marcapasos; MSC: muerte súbita cardiaca; NYHA: clase funcional de la New York Heart Association; RMC: resonancia magnética cardiovascular; TA: taquicardia auricular; TdF: tetralogía de Fallot; TGA: transposición de las grandes arterias; TRAV: taquicardia por reentrada auriculoventricular; TRIA: taquicardia por reentrada intraauricular; TRNAV: taquicardia por reentrada del nódulo auriculoventricular; TSV: taquicardia supraventricular; TV: taquicardia ventricular; VD: ventrículo derecho; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cConsiderando el amplio espectro de CCA con afección del VI que pueden diferir de las cardiopatías adquiridas, el mayor riesgo de complicaciones relacionadas con el DAI en las CCA y la escasez de datos sobre el beneficio de los DAI para la prevención primaria de la MSC en las CCA, parece apropiado que el enfoque sea personalizado.
dLos datos son escasos y los factores de riesgo pueden ser específicos de la lesión, incluida la TV no sostenida, NYHA II/III, insuficiencia grave de la válvula AV y QRS ancho ≥ 140ms (TGA).
La HP es un factor pronóstico importante en las CC42, que requiere una atención especial durante el embarazo43 y antes de la reparación cardiaca u otros tipos de cirugía mayor. Hasta hace poco, la HP se definía por un aumento ≥ 25mmHg en la PAP media medida de modo invasivo en reposo44. Si bien ahora este criterio se ha reducido a > 20mmHg45, la clasificación de HP precapilar requiere que exista también un aumento de la RVP ≥ 3 UW (véase la tabla 8)45. El aumento de la resistencia en este grupo de pacientes se debe a una vasculopatía pulmonar obstructiva relacionada con antecedentes genéticos, genes modificadores, estrés de cizallamiento vascular y desencadenantes ambientales44. Es importante diferenciar entre la HP precapilar relacionada con CC del grupo 1 de la clasificación de HP44 y otras condiciones que cursan con aumento de la presión de llenado del VI > 15mmHg (HP poscapilar por transmisión pasiva de las presiones de llenado del VI de los grupos 2 y 5 de la clasificación)44, debido a que los tratamientos dirigidos a la HP poscapilar no funcionan. Los subtipos clínicos de las CC con HP precapilar son la HP precapilar con cortocircuitos sistémico-pulmonares congénitos, el síndrome de Eisenmenger, las malformaciones reparadas y la HP precapilar concomitante con CC, normalmente defectos pequeños. La circulación de Fontan es otra condición que se asocia con enfermedad vascular pulmonar (EVP) y ocasionalmente con RVP. No obstante, el aumento de la PAP en estos pacientes suele ser poscapilar (aumento de la presión de llenado o insuficiencia de válvula AV). En las CC complejas, la PH precapilar puede estar restringida a ciertos segmentos del lecho vascular pulmonar (HP precapilar segmentaria)46, sobre todo en casos complejos de atresia pulmonar con DSV.
Definiciones de los subtipos de hipertensión pulmonar y su aparición en las CCA
| Hipertensión pulmonar en la cardiopatía congénita del adulto | ||
|---|---|---|
| Definición | Características hemodinámicas* | Contextos clínicos |
| Hipertensión pulmonar (HP) | PAP media > 20 mmHg | Todos |
| HP precapilar (HAP) | PAP media > 20 mmHg PEAP ≤ 15 mmHg | Cortocircuitos antes y después de la reparación (incluido el síndrome de Eisenmenger) |
| RVP ≥ 3 WU | CC compleja (incluidos el CUV y la HAP segmentaria) | |
| HP poscapilar aislada | PAP media > 20 mmHg | Disfunción ventricular sistémica |
| PEAP > 15 mmHg | Disfunción de la válvula AV sistémica | |
| RVP < 3 WU | Obstrucción de la vena pulmonarCor triatriatum | |
| HP precapilar y poscapilar combinada | PAP media > 20 mmHgPEAP > 15 mmHg | Los contextos enumerados en la HP poscapilar aislada |
| RVP ≥ 3 WU | Los contextos enumerados en la HP poscapilar aislada en combinación con cortocircuitos/CC compleja | |
AV: auriculoventricular; CC: cardiopatía congénita; CCA: cardiopatías congénitas del adulto; CUV: corazón univentricular; HAP: hipertensión arterial pulmonar; HP: hipertensión pulmonar; PAP: presión arterial pulmonar; PEAP: presión de enclavamiento de la arteria pulmonar; RVP: resistencia vascular pulmonar.
*La definición más reciente de HP45 reduce la PAP media de ≥ 25 mmHg44 a > 20mmHg, pero se requiere además una RVP ≥ 3 WU para la HP precapilar.
La HP precapilar es más frecuentemente en mujeres, aunque puede aparecer en ambos sexos y a cualquier edad, y su prevalencia aumenta con la edad biológica y la edad al cierre del defecto47. El sesgo por sexos de la CC con HP precapilar desaparece después de reparar el defecto48. Recientemente, un estudio nacional sobre población con CC ha descrito una prevalencia de HP precapilar del 3,2% de los pacientes con CC, lo que representa 100 casos/millón de habitantes de la población general adulta47.
3.4.3.2DiagnósticoEn la guía de 2015 sobre el diagnóstico y el tratamiento de la HP, se presentaba un algoritmo hemodinámico para el diagnóstico de HP que resaltaba la importancia del cateterismo derecho para diferenciar la HP precapilar de la poscapilar44. Este algoritmo se ha modificado recientemente45. La tabla 8 ilustra las definiciones actuales de los diferentes tipos de HP y los contextos en que pueden producirse en pacientes con CCA. Según esta nueva definición, el umbral medio de PAP debe ser > 20, en lugar de ≥ 25mmHg, en presencia de una RVP ≥ 3 UW para cumplir los criterios de HP precapilar45.
3.4.3.2.1. Estudio diagnóstico de la hipertensión pulmonar en las cardiopatías congénitas del adulto. El estudio diagnóstico incluye la historia clínica, exploración física, pruebas de función pulmonar, análisis de gases en sangre arterial, estudio por imagen (especialmente ecocardiográfica) y pruebas de laboratorio (recuento total de células sanguíneas, concentración sérica de hierro, hematocrito, marcadores de infección y determinaciones de NT-proBNP). En general, se requiere cateterismo del corazón derecho con oximetría para la toma de decisiones mayores, como el inicio y el seguimiento del tratamiento vasodilatador, la posibilidad de embarazo y la cirugía. El umbral para la evaluación invasiva es más alto en pacientes con Eisenmenger. No suele ser necesaria la evaluación hemodinámica invasiva para guiar las intervenciones terapéuticas a lo largo del tiempo. Se deben tener en cuenta los valores de hematocrito, ya que cuando son altos aumentan la RVP49.
3.4.3.2.2. Evaluación del riesgo. Los resultados clínicos de los pacientes con HP precapilar han mejorado gracias a los nuevos tratamientos, los avances en el tratamiento quirúrgico y perioperatorio y las estrategias multidisciplinarias con equipos médicos especializados44,50,51. En las series recientes, este grupo de pacientes parece ir mejor que el grupo con HP idiopática48, aunque puede variar dependiendo del subtipo de HP. Los resultados clínicos de la HAP asociada con defectos pequeños son tan pobres como los de la HAP idiopática, probablemente porque ambas condiciones comparten el mismo tipo de anomalía proliferativa vascular. La HAP tras la reparación tiene un pronóstico aún peor48.
3.4.3.3Tratamiento de la hipertensión pulmonar en las cardiopatías congénitas del adulto3.4.3.3.1. Centros con experiencia.
Para tratar con éxito a un paciente con CCA e HP, se requiere un equipo multidisciplinario compuesto por expertos en imagen, cardiología, medicina respiratoria, hematología, enfermedades infecciosas, obstetricia, anestesiología, neonatología, HP, cirugía torácica y cardiovascular, enfermería y genética médica.
3.4.3.3.2. Medidas generales. Las principales medidas generales son el apoyo social y psicológico, la vacunación y evitar el estrés físico excesivo. Las visitas de seguimiento deben planificarse individualizadamente. Debe evitarse el embarazo en todos los casos de HP precapilar. Se recomienda oxígeno suplementario continuo cuando la presión arterial de oxígeno sea persistentemente < 60mmHg44, excepto para los pacientes con Eisenmenger, que solo lo tienen recomendado cuando esta maniobra produce un aumento documentado, constante y significativo de la saturación de oxígeno y una mejoría de los síntomas.
3.4.3.3.3. Anticoagulación. En general, la anticoagulación con antagonistas de la vitamina K en ausencia de arritmia auricular, válvulas mecánicas o prótesis vascular no está recomendada en las CC con HAP, y su prescripción debe ser individualizada, por ejemplo, en caso de arterias pulmonares (AP) grandes y aneurismáticas con presencia de trombo o cuando se hayan producido episodios tromboembólicos previos. No hay datos sobre los nuevos anticoagulantes orales no antagonistas de la vitamina K (NACO). Para los pacientes con Eisenmenger, se deben considerar los anticoagulantes orales en caso de arritmias auriculares. También pueden ser necesarios cuando haya trombosis de la AP o embolización, siempre que el riesgo hemorrágico sea bajo. No obstante, no hay datos que respalden un uso sistemático. Debido a que el riesgo hemorrágico se encuentra aumentado en los pacientes cianóticos, es preciso evaluar cuidadosa e individualizadamente la prescripción de anticoagulantes y antiagregantes plaquetarios en este grupo de pacientes.
3.4.3.3.4. Reparación del cortocircuito. La reparación quirúrgica o mediante cateterismo intervencionista en las condiciones que cursan con aumento del flujo tienen la finalidad de proteger la vasculatura pulmonar, ya que el cizallamiento endotelial desencadena HP52. El umbral de la RVP que permite el cierre quirúrgico del cortocircuito izquierda-derecha (I-D) sin que se produzca insuficiencia cardiaca derecha difiere según el tipo de cortocircuito (véase los apartados 4.1-4.4). No obstante, la decisión de cerrar el cortocircuito debe tomarse a partir de toda la información disponible y no solo por la información hemodinámica obtenida durante el cateterismo cardiaco53,54. Solo los centros especializados pueden hacer esta valoración.
No hay evidencia prospectiva de que las estrategias de tratamiento y reparación en los pacientes con HAP asociada con CC confieran beneficios a largo plazo44,55. Tampoco hay datos prospectivos sobre la utilidad de las pruebas de vasorreactividad, la prueba de cierre o la biopsia pulmonar para evaluar la idoneidad de la cirugía53,54,56–60.
3.4.3.3.5. Tratamiento médico para la hipertensión arterial pulmonar. Los tratamientos avanzados benefician a los pacientes con Eisenmenger51 y probablemente a pacientes con otras CC asociadas con HAP61,62. Según la guía de la ESC de 2015 sobre HP44, la HP precapilar, incluido el síndrome de Eisenmenger, es una condición de riesgo moderado a alto que requiere un enfoque proactivo con tratamiento inicial combinado63 o secuencial64,65, que incluya la administración de prostaciclinas parenterales44. Las prostaglandinas parenterales funcionan mejor cuando se inician pronto66. No obstante, la presencia de un catéter intravenoso central para el tratamiento parenteral aumenta el riesgo de embolización paradójica e infección en pacientes con Eisenmenger o cortocircuito D-I. Por consiguiente, son preferibles las vías de administración subcutánea o inhalada. Las excepciones a esta regla son los pacientes con defectos cerrados o coincidentes que cumplan criterios estrictos de respuesta vasodilatadora (principalmente una disminución aguda de la PAP media > 10mmHg y por debajo de 40mmHg con óxido nítrico inhalado) y solo puedan ser tratados con antagonistas de los canales de calcio. Estos pacientes son extremadamente raros entre los adultos con HAP asociada con CC. No se recomienda la prueba de vasorreactividad general en la HAP asociada con CC44. La oxigenoterapia domiciliaria de larga duración puede mejorar los síntomas, pero no se ha demostrado que aumente la supervivencia de los pacientes con Eisenmenger. Los suplementos de oxígeno deben restringirse a casos en los que el tratamiento produzca un aumento documentado, constante y significativo de la saturación de oxígeno arterial y mejoría de los síntomas. La eritrocitosis secundaria es beneficiosa para el transporte y la liberación de oxígeno y se debe evitar la flebotomía sistemática (véase el apartado 3.4.8.). En pacientes con Eisenmenger en clase funcional III de la Organización Mundial de la Salud (OMS), se ha demostrado que el antagonista del receptor de la endotelina (ARE) bosentán mejora la PM6M y reduce la RVP después de 16 semanas de tratamiento67. Aunque se ha descrito un efecto beneficioso del bosantán en la capacidad de esfuerzo y la calidad de vida de este grupo de pacientes, el efecto en la mortalidad es más incierto. El seguimiento a largo plazo ha demostrado una persistente mejoría de los síntomas. La experiencia con otros ARE y con los inhibidores de la fosfodiesterasa 5 (FDE-5) sildenafilo y tadalafilo también ha arrojado resultados funcionales y hemodinámicos favorables en pacientes con HAP asociada con CC y síndrome de Eisenmenger, aunque la evidencia es menos robusta. La experiencia terapéutica que se tiene en la HAP asociada con CC se limita a los fármacos de última generación, como el macitentán, el selexipag y el riociguat66. Si bien se ha incluido a los pacientes con lesiones reparadas simples en los estudios de referencia para estos fármacos, y es probable que se beneficien de manera similar que los pacientes con HAP idiopática, solo se dispone de datos de pacientes con sídrome de Eisenmenger. Los resultados de un reciente ensayo controlado aleatorizado sobre la eficacia del macitentán para mejorar la PM6M en el síndrome de Eisenmenger han sido neutrales68. La mayoría de los centros siguen una estrategia de tratamiento secuencial orientada a los síntomas en el síndrome de Eisenmenger, que empieza generalmente con un ARE o un inhibidor de la PDE–5 oral y se continúa con un tratamiento escalonado cuando los síntomas persisten o hay deterioro clínico. Si no se logra una mejoría adecuada de los síntomas con tratamiento oral, deben considerarse las opciones parenterales de manera proactiva. El efecto del tratamiento de la HAP en pacientes con HAP segmentaria sigue siendo controvertido. Aunque algunas series han descrito resultados prometedores, ha habido casos en los que el tratamiento no se ha tolerado46. El trasplante cardiopulmonar o el trasplante de pulmón con cirugía cardiaca es una opción en casos especiales que no responden al tratamiento médico, pero se encuentra limitada por la complejidad quirúrgica y la disponibilidad de órganos.
Recomendaciones para el tratamiento de la hipertensión arterial pulmonar asociada con cardiopatía congénita
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Se recomienda desaconsejar el embarazo a las pacientes con CC e HP precapilar confirmadac | I | C |
| Se recomienda la evaluación del riesgo de todos los pacientes con HAP-CCd | I | C |
| Para los pacientes con riesgo bajo e intermedio63–65, lesiones simples reparadas y HP precapilar, se recomienda el tratamiento oral combinado inicial o combinado secuencial; se debe tratar a los pacientes en alto riesgo con tratamiento combinado inicial que incluya prostanoides parenteralese | I | A |
| Para los pacientes con síndrome de Eisenmenger y capacidad de ejercicio reducida (distancia en la PM6M < 450 m), se debe considerar una estrategia de monoterapia inicial con un antagonista del receptor de la endotelina seguida de tratamiento combinado si los pacientes no mejoran67–69 | IIa | B |
CC: cardiopatía congénita; HAP-CC: hipertensión arterial pulmonar asociada con cardiopatía congénita; HP: hipertensión pulmonar; PM6M: prueba de los 6min de marchA.
aClase de recomendación.
bNivel de evidencia.
cEl riesgo del embarazo para las pacientes con HP precapilar es muy elevado. Puede ser menor para las pacientes con HP poscapilar, por lo que para confirmar el diagnóstico es necesario un cateterismo cardiaco derecho para toda paciente con sospecha de HP precapilar.
dPara conocer más detalles, véase la guía de la ESC/ERS de 2015 sobre diagnóstico y tratamiento de la HP44.
ePara conocer más detalles sobre la elección de los fármacos y el algoritmo del tratamiento ajustado al riesgo, véase la guía de la ESC/ERS de 2015 sobre diagnóstico y tratamiento de la HP44.
La cirugía cardiaca merece una atención especial en los pacientes con CCA. Incluso las intervenciones pequeñas conllevan un riesgo alto, ya que pueden alterar el equlibrio delicado en el que suelen encontrarse estos pacientes. No es posible calcular este riesgo mediante los sistemas convencionales de puntuación de riesgo quirúrgico. El sistema de puntuación de la cirugía de CCA procede de la base de datos de la Society of Thoracic Surgeons Congenital Heart Surgery, que fue el primer sistema basado en la evidencia específicamente diseñado para la cirugía de las CCA70.
La evaluación de este sistema de puntuación demostró que tenía un buen poder predictivo en las CCA, mientras que en los niños funcionaba mejor el sistema de puntuación pediátrica71, lo que indica que las comorbilidades de los pacientes individuales tienen un papel importante en la predicción de los resultados clínicos. Por lo tanto, la puntuación del riesgo de mortalidad en las CCA, una puntuación basada en la opinión de expertos que tiene en cuenta las comorbilidades, también puede considerarse para evaluar el riesgo de la cirugía cardiaca en estos pacientes72.
Además de la evaluación del riesgo personalizada, hay otros factores que pueden determinar los resultados a corto y largo plazo, como la adecuada comprensión de la anatomía y la hemodinámica específicas, la experiencia en la repetición de la cirugía y los requisitos específicos en las unidades de cuidados intensivos. Se ha demostrado que cuando los pacientes con CCA son operados por cirujanos expertos en cardiopatías congénitas, los resultados son superiores73. Por consiguiente, la recomendación estricta es que todos los pacientes con CCA sean operados por cirujanos expertos en cardiopatías congénitas dentro de un entorno multidisciplinario con experiencia en CCA. Esta recomendación es válida para toda la cirugía cardiaca en el contexto de las CCA, excepto para la cirugía de la válvula aórtica bicúspide (VAB) no complicada, la enfermedad hereditaria de la aórtica torácica (EHAT), como el síndrome de Marfan, y la CIA de tipo ostium secundum sin conexión venosa pulmonar anómala o ausencia de EVP. El entorno de expertos multidisciplinarios fomenta un interés creciente por desarrollar procedimientos híbridos, en los que deben colaborar cirujanos cardiacos (de CCA), cirujanos vasculares, cardiólogos intervencionistas en CCA y electrofisiólogos para llevar a cabo procedimientos que representan un reto técnico.
3.4.5Cateterismo intervencionistaEl cateterismo, ya sea como procedimiento independiente o híbrido, es una alternativa atractiva a la cirugía convencional a corazón abierto, ya que evita la necesidad de repetir la esternotomía/toracotomía y la derivación cardiopulmonar. Las intervenciones percutáneas más frecuentes son el cierre de cortocircuitos (especialmente la CIA de tipo ostium secundum, más raramente los DSV y el ductus arterial persistente [DAP]), fístulas o colaterales atípicas; la dilatación con balón de la válvula pulmonar e injertos valvulados; la dilatación con balón o colocación de stents en las grandes arterias estenosadas (p. ej., nueva coartación aórtica [CoA] y estenosis arterial pulmonar), y el implante percutáneo de la válvula pulmonar (IPVP). El diagnóstico de CCA y, en particular, la atención durante el intervencionismo deben correr a cargo de personas capacitadas como cuidadores en CC y que formen parte de un centro de atención de CCA en el que los procedimientos individuales se revisan y discuten dentro de un equipo multidisciplinario74. Es muy importante que los programas de intervención percutánea de pacientes con CCA se ubiquen junto con los servicios de CC correspondientes para el abordaje de las complicaciones de los procedimientos. En muchos programas, los cardiólogos intervencionistas pediátricos con experiencia son un componente importante de la unidad de intervencionismo de las CCA, pero en otros (como los que se encuentran en un hospital pediátrico independiente), los intervencionistas pediátricos pueden sentirse incómodos al tener que tratar a adultos. Por lo tanto, se recomienda la plena colaboración entre la cardiología de adultos y la pediátrica. La centralización del cateterismo de los pacientes con CCA en un pequeño número de centros permite garantizar la calidad y la disponibilidad de los servicios cardiovasculares esenciales74. Recientemente se ha propuesto un número mínimo de intervenciones anuales por centro74.
3.4.6Endocarditis infecciosaEl riesgo de endocarditis infecciosa (EI) de los pacientes con CCA es mayor que para la población general, y varía significativamente según el tipo de lesión. La guía de la ESC de 2015 sobre EI mantiene la restricción de la profilaxis antibiótica a los pacientes con riesgo alto de EI que se someten a procedimientos dentales de riesgo22. Las condiciones de riesgo alto son la presencia de válvulas protésicas, incluidas las de implante percutáneo, la reparación valvular con anillo protésico, la EI previa, cualquier CC cianótica y cualquier CC reparada con material protésico hasta 6 meses después del procedimiento o de por vida si persiste un cortocircuito residual o insuficiencia valvular.
Deben aplicarse medidas higiénicas no específicas para todos los pacientes con CCA: adecuada higiene bucal y cutánea, y medidas asépticas durante la atención sanitaria y el procedimiento invasivo. Se desaconsejan los piercings y los tatuajes y, en caso de hacerse, deben cumplirse las condiciones higiénicas óptimas.
Se debe asesorar a todos los pacientes sobre los síntomas de EI y cómo actuar en el momento de su aparición (buscar consejo médico, importancia de los hemocultivos antes de iniciar el tratamiento antibiótico).
Estudios recientes han confirmado el riesgo relativamente alto de EI de los pacientes que se han sometido a cirugía valvular, sobre todo en casos de EI previa y en portadores de conductos yugulares bovinos75-78. Se debe prestar una atención especial después de una IPVP con la prótesis valvular Melody75.
3.4.7Tratamiento antitrombóticoLos pacientes con CCA tienen mayor riesgo de sufrir eventos tromboembólicos, pero hay poca evidencia sobre su prevención. En los casos de TRIA o FA, se ha demostrado el beneficio de emplear las escalas CHA2DS2-VASc y HAS-BLED en las CCA38. No obstante, dado que su validez en la población con CC en general no se conoce, estos sistemas de puntuación deben combinarse con la evaluación del riesgo individual. Tradicionalmente, los antagonistas de la vitamina K se han utilizado para la prevención tromboembólica, aunque en cardiología general se prefieren los NACO. Los NACO también son seguros y eficaces en la población con CCA en ausencia de válvulas mecánicas o estenosis mitral grave79,80. Se recomienda el tratamiento anticoagulante en la FA/TRIA paroxística y persistente de los pacientes con CC moderada o grave, pero es necesario aplicar una estrategia individualizada. En pacientes con CCA leve, las escalas CHA2DS2-VASc y HAS-BLED son útiles para las recomendaciones generales81. Se desconoce por el momento si el tratamiento anticoagulante beneficia a todos los pacientes con circulación de Fontan. Es importante sopesar el riesgo hemorrágico frente al riesgo trombogénico, sobre todo en pacientes cianóticos. Se recomienda el tratamiento anticoagulante en la prevención secundaria de pacientes con un evento tromboembólico o un trombo intracardiaco o intravascular incidental (véase también la guía de la ESC/EACTS de 2017 sobre el tratamiento de la cardiopatía valvular y la guía de la ESC de 2018 sobre embarazo).
3.4.8Tratamiento de los pacientes cianóticosLa cianosis está causada por un cortocircuito D-I debido a una comunicación anatómica entre la circulación sistémica y la pulmonar en la aurícula, el ventrículo o las arterias. La cardiopatía cianótica comprende un grupo heterogéneo de lesiones con diferentes fisiopatología y anatomía subyacentes: puede haber flujo sanguíneo pulmonar normal o restringido en presencia de una obstrucción a través del tracto pulmonar de salida o flujo sanguíneo pulmonar aumentado en ausencia de obstrucción que, en algunos defectos, puede llevar a la aparición de HAP y eventualmente síndrome de Eisenmenger (véase los apartados 3.4.3 y 4.15). Estos casos pueden presentarse con o sin intervención paliativa previa. Los cianóticos son pacientes complejos y su seguimiento debe llevarse a cabo por un especialista en CCA.
3.4.8.1Mecanismos adaptativosLa cianosis induce la aparición de mecanismos adaptativos para mejorar el transporte de oxígeno y su liberación a los tejidos: eritrocitosis secundaria, desplazamiento hacia la derecha de la curva de disociación de la oxihemoglobina y aumento del gasto cardiaco82,83. La eritrocitosis secundaria al estímulo de la eritropoyetina es la respuesta fisiológica a la hipoxemia crónica. La eritrocitosis compensada refleja un estado de equilibrio, mientras que la eritrocitosis descompensada indica que el equilibrio no se puede mantener (aumento excesivo de eritrocitos/hemoglobina y hematocrito inestable y creciente con síntomas de hiperviscosidad)82,84.
3.4.8.2Trastorno multisistémicoLa cianosis y la eritrocitosis secundaria implican profundas consecuencias en todo el sistema orgánico82,84,85:
- •
–La viscosidad sanguínea aumenta en relación directa con la masa de eritrocitos84.
- •
Las anomalías hemostáticas son comunes y complejas, y se atribuyen a anomalías en las plaquetas (trombocitopenia y trombastenia), vías de coagulación y otros mecanismos anómalos de coagulación. Los factores de coagulación dependientes de la vitamina K (factores II, VII, IX y X) y el factor V disminuyen, aumenta la actividad fibrinolítica y se agotan los multímeros más grandes del factor de von Willebrand.
- •
La mayor recuperación de eritrocitos/hemoglobina y la filtración de ácido úrico deteriorada causan hiperuricemia86. La mayor concentración de bilirrubina no conjugada pone a los cianóticos en riesgo de cálculo biliar de bilirrubinato de calcio.
- •
La disfunción endotelial grave es evidente por el llamativo deterioro de la vasodilatación dependiente del endotelio85.
- •
La hipoxemia crónica, la mayor viscosidad sanguínea y la disfunción endotelial afectan a la microcirculación, la función miocárdica y la función de otros sistemas orgánicos.
La presentación clínica incluye cianosis central, como resultado de la mayor cantidad de hemoglobina reducida (> 5 g/100ml de sangre), acropaquia y, a menudo, escoliosis. Los hallazgos clínicos varían y se caracterizan por la anatomía/fisiopatología de base.
La mortalidad de los cianóticos es mucho mayor que la de los no cianóticos87. El resultado está determinado por la anatomía, la fisiopatología, las intervenciones paliativas, las complicaciones de la cianosis y las medidas preventivas de base88,89. Un recuento plaquetario bajo, hipoxia grave, cardiomegalia y hematocrito elevado durante la infancia son parámetros útiles para predecir la muerte prematura y los efectos secundarios en pacientes con o sin EVP90. La deficiencia de hierro se asocia con un resultado tardío adverso91,92. El valor de BNP puede predecir el resultado clínico en pacientes con Eisenmenger93,94. No obstante, en un estudio multicéntrico89, solo la edad, el cortocircuito pretricuspídeo, la ausencia de ritmo sinusal, la saturación de oxígeno baja en reposo y la efusión pericárdica, pero no el BNP, fueron los más potentes factores predictivos de muerte.
3.4.8.4Complicaciones tardías- •
Los síntomas de hiperviscosidad incluyen cefaleas, desmayos, mareos, fatiga, tinnitus, visión borrosa, parestesia de labios y dedos de manos y pies, dolor muscular y debilidad, clasificados como moderados cuando interfieren con algunas actividades y como graves cuando interfieren con la mayoría de las actividades82,83. Es poco probable que un paciente con reservas normales de hierro y hematocrito < 65% tenga síntomas de hiperviscosidad.
- •
Las diátesis hemorrágicas y trombóticas ocurren, y ambas originan un dilema terapéutico, ya que los pacientes tienen riesgo de trombosis y hemorragia. La hemorragia espontánea suele ser menor y autolimitada (hemorragia dental, epistaxis, formación fácil de hematomas y menorragia). La hemoptisis es el evento hemorrágico grave más común y la manifestación externa de una hemorragia intrapulmonar que no refleja el alcance de la hemorragia del parénquima (constatada en hasta el 100% de los pacientes con Eisenmenger)95,96. La trombosis es consecuencia de anomalías en la coagulación, estasis sanguínea en las cámaras y vasos dilatados, ateroesclerosis o disfunción endotelial, material trombogénico en los conductos y arritmias. Las anomalías hemostáticas no protegen de las complicaciones trombóticas. Los trombos laminares de arterias pulmonares grandes aneurismáticas y parcialmente calcificadas son comunes (hasta el 30%)97–100. El sexo femenino, una baja saturación de oxígeno, la edad avanzada, la disfunción biventricular y las AP dilatadas se han definido como factores de riesgo97,100,101.
- •
Los infartos cerebrovasculares son frecuentes, pero están infradocumentados97. Podrían deberse a eventos tromboembólicos (émbolos paradójicos, arritmia supraventricular), factores reológicos (microcitosis), disfunción endotelial y factores de riesgo atereosclerótico «tradicionales». La gravedad de la eritrocitosis secundaria per se no es un factor de riesgo102. La microcitosis causada por déficit de hierro debido a flebotomías inapropiadas es el más importante factor independiente predictivo de eventos cerebrovasculares91. Otros factores de riesgo son la gravedad de la cianosis y la complejidad de la CC97.
- •
Los émbolos paradójicos podrían ser consecuencia de derivaciones transvenosas o catéteres.
- •
El déficit de hierro suele ser consecuencia de flebotomías inapropiadas o menstruaciones abundantes en mujeres.
- •
Arritmias supraventriculares y ventriculares.
- •
Las complicaciones infecciosas incluyen endocarditis, abscesos cerebrales y neumonía. La fiebre secundaria a cefaleas nuevas o diferentes levanta sospecha de abscesos cerebrales.
- •
La disfunción renal es frecuente y consecuencia de anomalías funcionales y estructurales de los riñones.
- •
La colelitiasis es común. Los cálculos biliares de bilirrubinato de calcio pueden complicarse por colecistitis/coledocolitiasis.
- •
Las complicaciones reumáticas incluyen artritis gotosa, osteoartropatía hipertrófica y quifoscoliosis83.
Debe prestarse especial atención a los síntomas de hiperviscosidad, así como a las complicaciones hemorrágicas/isquémicas. La saturación de oxígeno debe obtenerse mediante oximetría de pulso en reposo ≥ 5min, y la capacidad de ejercicio debe evaluarse con regularidad, preferiblemente con PM6M.
Los análisis de sangre deben incluir hemograma, volumen corpuscular medio (VCM), ferritina en suero (podría ser necesario determinar en suero hierro, transferrina y saturación de transferrina para la detección precoz de déficit de hierro), creatinina, ácido úrico en suero, perfil de coagulación, BNP o NT-proBNP, ácido fólico y vitamina B12 en presencia de VCM elevado o normal y ferritina en suero baja.
3.4.8.6Precauciones de laboratorio–
- •
Parámetros de coagulación: el volumen de plasma se reduce por la eritrocitosis secundaria; la cantidad de citrato sódico debe ajustarse por hematocrito si este es > 55%.
- •
El hematocrito se determina con recuentos automatizados de partículas eléctricas (la centrífuga de microhematocrito resulta en un hematocrito falsamente alto por el atrapamiento plasmático).
- •
La glucosa puede reducirse debido a un aumento de la glucolisis in vitro como consecuencia del mayor número de eritrocitos.
Los riesgos y beneficios deben considerarse cuidadosamente y requieren experiencia. Un paciente cianótico sin HAP/síndrome de Eisenmenger debe ser evaluado periódicamente por cualquier intervención que pudiera mejorar la calidad de vida y reducir la morbilidad o por elegibilidad para reparación fisiológica (véase el apartado 4.15).
3.4.8.8Tratamiento médico- •
Para el tratamiento específico de la HAP, véase el apartado 3.4.3.
- •
Arritmias: el ritmo sinusal debe mantenerse siempre que sea posible. El tratamiento antirrítmico debe ser individualizado (fármacos, ablación, MP/DAI epicárdico) y es extremadamente difícil en este grupo de pacientes. El tratamiento farmacológico debe instaurarse con cuidados especiales y, en general, en un hospital.
- •
La flebotomía terapéutica debe realizarse únicamente en presencia de síntomas de hiperviscosidad moderados/graves debidos a eritrocitosis secundaria (hematocrito > 65%) y en ausencia de deshidratación y déficit de hierro82. Debe reemplazarse el fluido isovolumétrico (750-1.000ml de solución salina isotónica mientras se extraen 400-500ml de sangre).
- •
La transfusión de sangre puede ser necesaria en caso de anemia ferropénica (volumen de hemoglobina inadecuado para la saturación de oxígeno) siguiendo las indicaciones convencionales.
- •
Deben administrarse suplementos de hierro en caso de déficit de hierro (VCM < 80 fl, reservas de hierro bajas), con cuidadoso seguimiento (efecto de repunte).
- •
Anticoagulación/ácido acetilsalicílico sistemáticamente: los datos disponibles hoy no avalan que prevenir las complicaciones tromboembólicas sea beneficioso para los cianóticos. Sin embargo, hay mayor riesgo de hemorragia.
- •
Indicación para la anticoagulación: flutter/FA (razón internacional normalizada [INR] 2-2,5; INR objetivo más alta en presencia de otros factores de riesgo, como válvula mecánica). Precauciones de laboratorio: pueden encontrarse valores de INR falsamente elevados debido al aumento del hematocrito. No hay resultados sobre los NACO (véase el apartado 3.4.7).
- •
Hemoptisis: exige radiografía torácica seguida de TC torácica si hay un infiltrado. La broncoscopia pone al paciente en riesgo y pocas veces proporciona información útil. El tratamiento incluye interrupción del ácido acetilsalicílico, los antiinflamatorios no esteroideos y los anticoagulantes orales, tratamiento contra la hipovolemia y la anemia, reducción de la actividad física y eliminación de la tos no productiva. La embolización selectiva de las arterias bronquiales puede ser necesaria por la hemorragia intrapulmonar refractaria/hemoptosis. Se están investigando fármacos antifibrinolíticos (como el ácido tranexámico inhalado) como estrategia novedosa para tratar la hemoptisis103. Se requieren más estudios clínicos.
- •
Hiperuricemia: no hay indicaciones para el tratamiento de la hiperuricemia asintomática.
- •
La artritis gotosa aguda se trata con colchicina oral o intravenosa, probenecid y fármacos antiinflamatorios, y hay que prestar especial atención al riesgo de insuficiencia renal y hemorragia. Los agentes uricosúricos, como el probenecid, o uricostáticos, como el alopurinol, evitan la recurrencia.
Todos los cianóticos precisan de por vida evaluación con visitas de seguimiento cada 6-12 meses en un centro especializado en CCA, con estrecha colaboración del médico de familia. La evaluación incluye:
- •
Evaluación completa y revisión sistemática de las complicaciones potenciales.
- •
Análisis de sangre (véase el apartado 3.4.8.8).
- •
Formación en estrategias de reducción del riesgo (tabla 9).
Tabla 9.Estrategias de reducción del riesgo de los pacientes con cardiopatía congénita cianótica
Las medidas profilácticas son el pilar del cuidado para evitar complicaciones83 Deben evitarse las siguientes exposiciones/actividades: Otras estrategias de reducción del riesgo: • Embarazo de pacientes con síndrome de Eisenmenger y pacientes cianóticas sin HAP pero con saturación arterial de oxígeno < 90% • Uso de un filtro de aire por vía intravenosa para prevenir la embolia gaseosa • Déficit de hierro y anemia (sin flebotomías sistemáticas inapropiadas para mantener una concentración de hemoglobina predeterminada): tratar el déficit de hierro y la anemia debida a deficiencia de hierro • Consulta con un cardiólogo de CCA antes de administrar cualquier fármaco o cualquier intervención quirúrgica/intervencionista • Anticoagulación inadecuada • Tratamiento antibiótico puntual contra las infecciones de las vías respiratorias superiores • Deshidratación • Uso prudente o evitación de agentes que deterioran la función renal • Enfermedad infecciosa: vacunación antigripal y antineumocócica • Consejo sobre anticoncepción en todas las visitas • Tabaquismo, abuso de drogas, incluido el alcohol • Cables MP/DAI transvenosos • Ejercicio intenso • Exposición a calor intenso (sauna, jacuzzi/ducha) o al frío extremo • Anticonceptivos con estrógenos CC: cardiopatía congénita; DAI: desfibrilador automático implantable; HAP: hipertensión arterial pulmonar; MP: marcapasos.
–
- •
Viajes aéreos: los vuelos comerciales se toleran bien104,105. Las estrategias de reducción del riesgo incluyen evitar el estrés, secundario o no a los viajes, la deshidratación y las bebidas alcohólicas, además de medidas para prevenir la trombosis venosa profunda.
- •
Exposición a altitudes elevadas: debe evitarse la exposición aguda a altitudes elevadas (> 2.500 m). El ascenso gradual, por ejemplo en teleférico, podría tolerase hasta los 2.500 m.
- •
Embarazo: en cianóticas sin HP, el embarazo tiene importantes complicaciones maternas y fetales y requiere el seguimiento de un equipo multidisciplinario. La saturación de oxígeno > 85% y la hemoglobina < 200g/l antes del embarazo son los principales factores predictivos de nacimiento vivo106. El embarazo está muy contraindicado para las mujeres con síndrome de Eisenmenger y las pacientes cianóticas sin HAP pero con una saturación de oxígeno < 90%43 (véase el apartado 3.5.7).
- •
Profilaxis contra la EI: se recomienda para todos los pacientes (véase el apartado 3.4.6).
Los ensayos clínicos han incluido un número suficiente de mujeres y, hasta cierto punto, se dispone de análisis estratificados por sexo. Los resultados sobre las diferencias entre sexos en la prevalencia y la morbimortalidad de las CC son controvertidos76,107-110. El estudio CONgenital CORvitia (CONCOR) no detectó diferencias en la mortalidad debidas al sexo, pero sí en la morbilidad: mayor riesgo de HP y menor riesgo de EI, complicaciones aórticas e implante de DAI en las mujeres111. Es necesario investigar si estas diferencias se deben a causas genéticas y biológicas inherentes al sexo, al menor tamaño corporal o a factores aún no identificados112.
Las diferencias por sexo son importantes para el diagnóstico y la toma de decisiones clínicas. Aunque las recomendaciones no suelen ser específicas para cada sexo, el uso de valores normalizados por el área de superficie corporal para establecer los puntos de corte de ciertos parámetros, como las dimensiones de la aorta y las cámaras cardiacas, puede corregir por la menor área de superficie corporal de las mujeres113. El asesoramiento personalizado es fundamental para las mujeres que planean gestar, ya que las indicaciones de intervención para las mujeres pueden basarse en valores absolutos más bajos en el caso de la cirugía aórtica (aortopatía hereditaria/asociada con CC) o en el reemplazo de la válvula pulmonar de las pacientes con TdF112.
Hay datos que muestran diferencias por sexo en el empleo114, la atención sanitaria115 y la actividad física116. Es conveniente adaptar la evaluación clínica, la toma de decisiones y el asesoramiento a las necesidades de cada persona para garantizar la equidad de los resultados clínicos.
3.5.2Cardiopatías congénitas del adulto a edades avanzadasActualmente, un ∼90% de los pacientes con CC leves, el 75% de los que las tienen moderadas y el 40% de los pacientes con CC complejas alcanzan los 60 años de edad117. Se espera que estos porcentajes aumenten en las próximas décadas. Por lo tanto, la población de personas mayores con CC que tiene unas necesidades únicas de atención médica está en aumento. Esta población de pacientes se caracteriza por tener más comorbilidades, riesgo inherente de arritmias relacionadas con la edad (especialmente FA), envejecimiento acelerado, enfermedad adquirida, cambios en las respuestas a la medicación, aparición más temprana de síndromes geriátricos (como deterioro cognitivo, inmovilidad/caídas, dificultades de desarrollo, alteraciones sensoriales), y un perfil psicosocial alterado118,119. Es fundamental consultar las guías específicas de CC en pacientes mayores para brindar una atención adecuada a este grupo de personas más vulnerables120. El inicio de enfermedades adquiridas comienza precozmente, por lo que las estrategias de prevención deben implementarse en las primeras décadas de vida (en cardiología pediátrica).
3.5.3Planificación anticipada de la atención y los cuidados paliativosLa mayoría de los pacientes, aparte de la complejidad de su CC, prefieren saber cuál es su esperanza de vida antes de enfrentarse a complicaciones potencialmente mortales121. Las complicaciones pueden ocurrir durante las intervenciones de riesgo o como consecuencia del incierto curso de la enfermedad. La planificación anticipada de la atención es un componente crítico de la atención integral centrada en el paciente121,122. Es difícil iniciar esta conversación; en este sentido, los ingresos hospitalarios no planificados, la inserción de un DAI o el deterioro funcional pueden ayudar. La mayoría de los pacientes prefieren esperar a que sea el médico quien inicie una conversación sobre la planificación anticipada de la atención. El contenido de estas conversaciones depende de la salud física y psicológica del paciente y de sus preferencias. Al principio, puede ser que el paciente solo desee recibir información sobre su esperanza de vida y las opciones terapéuticas. A medida que se deteriora la salud, es necesaria una revaluación integral de los deseos y los valores del paciente, las indicaciones anticipadas, la designación de un sustituto en la toma de decisiones y las decisiones con respecto a los dispositivos en pacientes con DAI.
Durante la atención médica, los cuidados paliativos pueden dar un apoyo progresivo al tratamiento específico de la enfermedad, hasta reemplazarlo totalmente. Sería aconsejable contar con la participación de especialistas en cuidados paliativos. En todo momento tiene que quedar claro que el tratamiento específico de la enfermedad puede continuar durante el proceso de planificación anticipada de la atención, incluso cuando se acerca el final de la vida, de acuerdo con las preferencias y los objetivos de los pacientes. La utilidad de los cuidados paliativos y el apoyo familiar continúa después de la muerte del paciente para la gestión del duelo123.
Siempre que sea posible, los miembros de la familia deben participar en todas las fases de la planificación. Las preferencias del paciente pueden cambiar con el tiempo y es necesaria una revaluación periódica de sus deseos.
3.5.4Seguro médico y empleoLos pacientes con CCA tienen dificultades para obtener seguros de vida, salud o viajes o un crédito hipotecario124–126. Si se otorga el seguro, los pacientes suelen tener que pagar un cargo adicional; a veces, el seguro médico puede excluir el tratamiento de la cardiopatía. En general, la disponibilidad del seguro y su tarifa no tienen relación con la complejidad de la enfermedad, el estado funcional del paciente o su pronóstico124,126, sino que dependen de las pólizas de las compañías de seguros, con sorprendentes diferencias entre países y dentro de ellos. Hoy en día los pacientes deben comparar precios, para lo que a veces las asociaciones de pacientes pueden ser de ayuda. En el futuro será necesario desarrollar estrategias nacionales uniformes para los seguros. Sin duda, la contratación de un seguro es un asunto importante que debe tratarse durante el asesoramiento de los pacientes. También es necesario prestar atención al empleo y los requisitos especiales de ciertas profesiones127. A partir de la adolescencia, las decisiones sobre la formación del paciente deben tomarse en función de sus posibilidades para realizar ejercicio intenso y turnos de noche y la toma de medicamentos específicos como los anticoagulantes orales.
3.5.5Ejercicio y deporteLas recomendaciones sobre ejercicio y deporte deben basarse en la capacidad del paciente, la cardiopatía congénita subyacente y el riesgo de complicaciones, el impacto en la hemodinámica de base y el riesgo de descompensación aguda y arritmias24. El asesoramiento debe basarse en el tipo de deporte y los niveles de esfuerzo previstos. La realización de las pruebas indicadas es inestimable y, en general, los médicos han sido demasiado conservadores en sus consejos. El ejercicio regular tiene beneficios bien documentados para la forma física, el bienestar psicológico y la interacción social, así como un efecto positivo en el futuro riesgo de cardiopatía adquirida; los síntomas no deben excluir la actividad física. Como recomendación general, el ejercicio dinámico es más adecuado que el estático.
En pacientes con enfermedades cardiacas conocidas, la muerte súbita durante el ejercicio es muy poco frecuente128. Las recomendaciones detalladas para la participación en deportes de competición van más allá del ámbito de este documento y se han publicado con anterioridad24. Las pautas sobre deportes de élite para atletas profesionales con CC se describen en la guía de la ESC de 2020 sobre cardiología deportiva129. La evaluación de la capacidad para hacer ejercicio ser previa a las recomendaciones sobre la práctica de una actividad recreativa o un deporte para evitar el ejercicio intenso a pacientes no entrenados. La mayoría de los pacientes con CC puede practicar con seguridad una actividad física moderada y regular. Algunas afecciones, como la disfunción sistólica del ventrículo sistémico, obstrucción del tracto de salida del ventrículo sistémico, HP, arritmias hemodinámicamente significativas o dilatación aórtica, requieren más precaución.
3.5.6Cirugía no cardiacaLa evaluación y el tratamiento de los pacientes con CCA deben seguir los principios de la guía de la ESC de 2014 sobre cirugía no cardiaca130 y tener en cuenta las especificidades de las CC. Los factores asociados con mayor riesgo de morbilidad y mortalidad perioperatoria son cianosis, insuficiencia cardiaca congestiva, mala salud general, edad más joven, HP, cirugía en los sistemas respiratorio y nervioso, CC compleja y procedimientos de urgencia/emergencia. La cirugía no cardiaca y los procedimientos intervencionistas de los pacientes con CC complejas (Fontan, Eisenmenger, pacientes cianóticos) deben llevarse a cabo en centros con experiencia130,131. Los aspectos que considerar son la profilaxis de la endocarditis, las complicaciones relacionadas con la hemodinámica de base, las anomalías en la anatomía venosa o arterial que afecten al acceso venoso y arterial, los cortocircuitos persistentes, las valvulopatías, las arritmias (incluidas las bradiarritmias), la eritrocitosis, la EVP, la prevención de la trombosis venosa, la monitorización de la función renal y hepática, la anticoagulación periprocedimiento, la posible necesidad de fármacos no convencionales y el aumento de la prevalencia de infección por hepatitis C debido a procedimientos previos y transfusiones de sangre.
3.5.7Embarazo, anticoncepción y asesoramiento genético3.5.7.1Embarazo y anticoncepciónLa mayoría de las pacientes con CCA toleran bien el embarazo, pero las mujeres con CCA complejas tienen un riesgo aumentado. En 2018 la ESC publicó una guía específica sobre embarazo y cardiopatías43.
Se debe ofrecer un asesoramiento previo al embarazo a todas las mujeres con CCA. Los cuidados especializados se aplican mejor dentro del marco de un equipo multidisciplinario. Este equipo debe contar con la colaboración de expertos en cardiología de CCA, ginecología, anestesia, hematología, neonatología y genética. El equipo debe participar tempranamente en el embarazo con el fin de planificar los cuidados prenatales, incluidos el parto y el seguimiento posparto. La valoración del riesgo debe ser individualizado de acuerdo con la clasificación de la OMS modificada (tabla 10)43.
Cardiopatía congénita con riesgo alto y extremadamente alto para el embarazo
| Riesgo significativamente mayor de muerte materna o morbilidad grave (clase III de la OMSm) (tasa de eventos cardiacos de un 19-27%) | Riesgo extremadamente alto de muerte materna o morbilidad grave (clase IV de la OMSm) (tasa de eventos cardiacos de un 40-100%) |
| Cardiopatía cianótica no reparada | Hipertensión arterial pulmonar |
| Disfunción del VI moderada (FE del 30-45%) | Disfunción del VI grave (FE < 30% o NYHA III-IV) |
| VD sistémico con función ventricular buena o ligeramente disminuida | VD sistémico con función ventricular moderada o disminuida grave |
| Circulación de Fontan. Si el paciente se encuentra bien y la afección cardiaca no presenta complicaciones | Fontan con alguna complicación |
| EA asintomática grave | EA sintomática grave |
| Estenosis mitral moderada | Estenosis mitral grave |
| Dilatación aórtica moderada (40-45 mm en síndrome de Marfan u otras EHAT; 45-50 mm en VAB, 20-25 mm/m2 en síndrome de Turner) | Dilatación aórtica grave (> 45 mm en síndrome de Marfan u otras EHAT, > 50 mm en VAB, > 25 mm/m2 en síndrome de Turner) |
| Válvula mecánica | (Re)coartación grave |
CC: cardiopatía congénita; EA: estenosis aórtica; EHAT: enfermedad hereditaria de la aorta torácica; FE: fracción de eyección; NYHA: clase funcional de la New York Heart Association; OMSm: escala de la Organización Mundial de la Salud modificada; TdF: tetralogía de Fallot; VAB: válvula aórtica bicúspide; VD: ventrículo derecho; VI: ventrículo izquierdo.
*Las mujeres con estas afecciones claramente deben evitar el embarazo.
Modificado de la guía de la ESC de 2018 sobre el tratamiento de las enfermedades cardiovasculares en el embarazo43.
El estado funcional antes del embarazo, la función ventricular, la gravedad de las lesiones y los antecedentes de eventos cardiacos son de especial valor pronóstico132. La PECP antes de la concepción puede predecir los resultados maternos y neonatales. Una respuesta de frecuencia cardiaca atenuada durante el ejercicio se asocia con mayor riesgo materno y neonatal de eventos cardiacos adversos.
La mortalidad materna es de un 0–1% y la insuficiencia cardiaca es una complicación del embarazo en el 11% de las mujeres con cardiopatías, y la HAP es la que conlleva mayor riesgo133,134. La cianosis implica un riesgo significativo para el feto, y el nacimiento vivo es poco probable (< 12%) cuando la saturación de oxígeno es < 85%106.
Las mujeres con cardiopatías también tienen mayor riesgo de complicaciones obstétricas, incluidos partos prematuros, preeclampsia y hemorragias posparto135. Siempre hay que tener en cuenta que los fármacos pueden afectar al feto. En concreto, no deben utilizarse inhibidores de la enzima de conversión de la angiotensina (IECA) ni antagonistas del receptor de la angiotensina II (ARA-II). Las mujeres que requieren anticoagulación oral merecen una atención especial. Los antagonistas de la vitamina K son teratogénicos, sobre todo a dosis alta. En la guía de la ESC de 2018 sobre embarazo se propone un algoritmo terapéutico ajustado por la dosis43.
La duración del embarazo y el modo de parto deben establecerse por un equipo multidisciplinario que tenga en cuenta la gravedad de la CC.
Se debe abordar la anticoncepción, con especial atención a su eficacia y su seguridad136. Los métodos de barrera son seguros y protegen contra enfermedades de transmisión sexual. No obstante, su eficacia anticonceptiva es alta solo en parejas estables. Los anticonceptivos hormonales son muy eficaces, pero hay pocos datos sobre su seguridad en la población con CCA. La anticoncepción oral combinada es muy eficaz (99,9%), pero conviene que la eviten las pacientes con riesgo trombótico preexistente (circulación de Fontan, cianosis, mala función ventricular sistémica), sobre todo porque hay pocos datos de que el tratamiento anticoagulante oral concomitante anule ese riesgo. Por otro lado, los anticonceptivos con solo progesterona no suponen un riesgo tan alto de trombosis, y los preparados orales más recientes tienen una eficacia alta, > 95%. El riesgo de EI tras la inserción de dispositivos intrauterinos impregnados en gestágenos probablemente sea bajo. No obstante, hay un riesgo de reacciones vasovagales del 5% en el momento de la inserción o retirada del dispositivo. La inserción/retirada del dispositivo intrauterino debe hacerse en un entorno seguro por profesionales con experiencia en CCA en casos de pacientes frágiles (circulación de Fontan, HP, cianosis, síndrome de Eisenmenger). La esterilización femenina o del compañero varón solo debería plantearse tras una cuidadosa discusión, con especial referencia al pronóstico a largo plazo.
Las técnicas de reproducción asistida conllevan otros riesgos aparte de los propios del embarazo; por ello, se debe consultar a un especialista en CCA antes de someterse a estos tratamientos. La superovulación es protrombótica y puede complicarse por el síndrome de hiperestimulación ovárica, que produce trastornos hemodinámicos importantes y un riesgo de trombosis aún mayor137. El riesgo de sufrir síndrome de hiperestimulación ovárica puede reducirse mediante monitorización cuidadosa del ciclo ovárico, hormonas estimuladoras del folículo a dosis baja combinadas con un antagonista de la gonadoliberina, congelación de todos los embriones y transferencia de un único embrión. Esta última opción está muy recomendada para las mujeres con cardiopatía, ya que los embarazos múltiples se asocian con cambios cardiovasculares más profundos y mayores complicaciones maternas y fetales138. El embarazo y los tratamientos de fertilidad están contraindicados para las mujeres con un riesgo cardiovascular materno de clase IV de la clasificación de la OMS modificada43. Las mujeres con riesgo cardiovascular de clase III o que están anticoaguladas tienen un riesgo muy alto de superovulación; en estos casos, se debe considerar la alternativa de la fertilización in vitro basada en el ciclo natural.
La sexualidad es importante en la calidad de vida. Los pocos datos disponibles apuntan a que la función sexual es motivo de preocupación tanto para las mujeres como para los varones y debería discutirse con más frecuencia139.
3.5.7.2Asesoramiento genético y riesgo de recurrenciaSe debe considerar el asesoramiento genético, ya sea acompañado de pruebas genéticas o no, para cada paciente con CCA. La demostración de una anomalía genética puede ser importante para ajustar el tratamiento del propio paciente y la planificación familiar. Se calcula que un 10–30% de todas las CC estructurales tienen una base genética. Esta tasa es mayor en los casos de anomalías orgánicas asociadas y casos familiares y menor en los casos aislados. Con el desarrollo técnico y las posibilidades que ofrecen las pruebas genéticas actuales, su fiabilidad ha mejorado mucho. El estudio genético debe ser multidisciplinario, debe integrarse a la información que aportan los datos clínicos y contar con una interpretación adecuada de las variantes genéticas encontradas. Recientemente se ha publicado sobre este tema un documento de consenso que proporciona un algoritmo para las pruebas genéticas y una descripción general de los principales síndromes que considerar140.
Un importante aspecto específico del asesoramiento genético es la evaluación del riesgo de recurrencia, que debe considerarse tanto en varones como en mujeres. La tasa de recurrencia de las CC en la descendencia varía del 2 al 50% y es mayor en mujeres que en varones. Los mayores riesgos de recurrencia se encuentran en los trastornos de un único gen o las anomalías cromosómicas, como Marfan, Noonan, síndrome de deleción de 22q11 y síndrome de Holt-Oram. En pacientes con solo una CC no familiar, la tasa de recurrencia varía del 1 al 21%141, dependiendo de la lesión sybyacente. En la tabla 11 se presenta una descripción general. Se recomienda la ecocardiografía fetal a las 19-22 semanas de embarazo cuando los progenitores estén afectados, aunque se puede realizar a las 15–16 semanas.
Tasas de recurrencia de diversas lesiones cardiacas congénitas según el sexo de los padres afectadosa
| Tasa de recurrencia (%)b | ||
|---|---|---|
| Mujeres | Varones | |
| CIA | 4-6 | 1,5-3,5 |
| DSV | 6-10 | 2-3,5 |
| DSAV | 11,5-14 | 1-4,5 |
| DAP | 3,5-4 | 2-2,5 |
| CoA | 4-6,5 | 2-3,5 |
| Marfan/EHAT | 50c | |
| OTSVI | 8-18 | 3-4 |
| OTSVD (EP) | 4-6,5 | 2-3,5 |
| Síndrome de Eisenmenger | 6 | NR |
| TdF | 2-2,5 | 1,5 |
| Atresia pulmonar/DSV | ND | ND |
| TGA | 2c | |
| TGAcc | 3-5c | |
| CUV (HLHS) | 21c | |
CC: cardiopatía congénita; CIA: comunicación interauricular; CoA: coartación aórtica; CUV: corazón univentricular; DAP: ductus arterioso persistente; DSAV: defecto del septo auriculoventricular; DSV: defecto del septo ventricular; EHAT: enfermedad hereditaria de la aorta torácica; EP: estenosis pulmonar; ND: no documentado; OTSVD: obstrucción del tracto de salida del ventrículo derecho; OTSVI: obstrucción del tracto de salida del ventrículo izquierdo; SHIH: síndrome del hemicardio izquierdo hipoplásico; TdF: tetralogía de Fallot; TGA: transposición de las grandes arterias; TGAcc: trasnsposición de las grandes arterias congénitamente corregida.
aModificado de Pierpont et al.141.
bExcepto para el síndrome de Marfan, las tasas se aplican a pacientes con lesiones cardiacas aisladas en los que se han excluido las entidades genéticas/sindrómicas conocidas.
cNo se dispone de datos específicos por sexo o no son relevantes.
- •
Es bastante común que la CIA permanezca sin diagnosticar hasta la edad adulta. Los tipos de CIA son:
- •
CIA del ostium secundum: el 80% de los CIA. Se localiza en la región del foramen oval y sus alrededores.
- •
CIA del ostium primum: el 15%. Sinónimos: defecto del septo auriculoventricular (DSAV), canal AV parcial. Se localiza cerca de la cruz cardiaca, las válvulas AV suelen tener malformaciones que resultan en varios grados de insuficiencia (véase el apartado 4.3).
- •
Defecto del seno venoso superior: el 5%. Se localiza cerca de la entrada de la vena cava superior (VCS), asociado con conexión parcial o completa de las venas pulmonares derechas con la VCS/AD.
- •
Defecto del seno venoso inferior: < 1%. Se localiza cerca de la entrada de la vena cava inferior (VCI).
- •
Seno coronario sin techo: < 1%. La separación de la aurícula izquierda (AI) puede faltar parcial o completamente.
Las lesiones secundarias incluyen conexión venosa pulmonar anómala, VCS izquierda persistente, estenosis de la válvula pulmonar y prolapso de la válvula mitral. Los defectos interauriculares son los más frecuentes en la anomalía de Ebstein (véase el apartado 4.9). Las decisiones terapéuticas de esta combinación son más complejas. El presente apartado aborda la CIA aislada.
El volumen del cortocircuito depende de la adaptabilidad del VD o el VI, el tamaño del defecto y la presión en la AI o la AD. La CIA simple resulta en cortocircuito I-D por la mayor adaptabilidad del VD que del VI (cortocircuito en general relevante, con un tamaño del defecto ≥ 10mm) y causa sobrecarga de volumen del VD y sobrecirculación pulmonar. La disminución de la adaptabilidad del VI o cualquier enfermedad que aumente la presión de la AI (hipertensión, cardiopatía isquémica, miocardiopatía, valvulopatía aórtica y mitral) aumenta el cortocircuito I-D. Como consecuencia, la CIA puede llegar a ser hemodinámicamente más importante con la edad. La menor adaptabilidad del VD (estenosis pulmonar, HAP u otras enfermedades del VD) o enfermedad de la válvula tricúspide pueden reducir el cortocircuito I-D o acabar causando su inversión, lo que produce cianosis.
4.1.2Presentación clínica e historia naturalLos pacientes suelen permanecer asintomáticos hasta la edad adulta; no obstante, pasada la cuarta década la mayoría tiene síntomas, como capacidad funcional reducida, disnea de esfuerzo y palpitaciones (taquiarritmias supraventriculares) y, con menor frecuencia, infecciones pulmonares e insuficiencia cardiaca derecha. La esperanza de vida se reduce en conjunto, pero la supervivencia es mucho mejor de lo que se suponía142. La PAP puede ser normal, pero por término medio aumenta con la edad. Sin embargo, la enfermedad vascular pulmonar grave es poco frecuente (< 5%) y se supone que su aparición requiere factores adicionales; el curso de la enfermedad es parecido al de la HAP idiopática48. Con el aumento de la edad y la PAP, las taquiarritmias se vuelven cada vez más comunes (flutter auricular y FA)143. La embolia sistémica podría ser consecuencia de la embolia paradójica, poco frecuente, o de la FA y el flutter auricular.
4.1.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos clave incluyen desdoblamiento del segundo ruido cardiaco y soplo sistólico del flujo pulmonar. Normalmente, el ECG revela bloqueo de rama derecha y desviación del eje a la derecha (desviación superior del eje a la izquierda en el DSAV parcial). La mayor vascularización pulmonar en la radiografía torácica suele pasar inadvertida.
- •
La ecocardiografía es la técnica diagnóstica clave, ya que ofrece el diagnóstico y la cuantificación. La sobrecarga de volumen del VD, que quizá sea el primer hallazgo inesperado en un paciente con CIA no diagnosticada antes, es el hallazgo clave y que mejor caracteriza la relevancia hemodinámica del defecto (preferible a la cuantificación del cortocircuito). En general, los defectos del seno venoso exigen un diagnóstico preciso por ETE (la RMC y la TCC son buenas alternativas y pueden ser superiores en caso de defectos del seno venoso inferior). La ETE también es necesaria para la evaluación precisa de los defectos del ostium secundum antes del cierre con dispositivo, que debería incluir dimensionamiento, exploración de la morfología del septo residual, tamaño y calidad del anillo, exclusión de otros defectos y confirmación de una conexión venosa pulmonar normal. La ecocardiografía 3D permite visualizar la morfología de la CIA. Otra información clave que obtener es la PAP y la IT.
- •
La RMC no suele ser necesaria, aunque puede ser útil para la evaluación de la sobrecarga de volumen del VD, la identificación del defecto del seno venoso inferior, la cuantificación del cociente de flujo pulmonar a sistémico (Qp:Qs) y la conexión venosa pulmonar (la alternativa para esta es la TCC).
- •
La cateterización cardiaca es necesaria en casos de PAP alta en ecocardiografía (PAP sistólica calculada > 40mmHg o signos indirectos cuando no pueda calcularse) para determinar la RVP.
- •
Los pacientes con HAP deben pasar una prueba de esfuerzo para excluir la desaturación.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención en la comunicación interauricular (nativa y residual)» y la figura 2.
 CIA: comunicación interauricular;
CIA: comunicación interauricular; Tratamiento de la comunicación interauricular. CIA: comunicación interauricular; HAP: hipertensión arterial pulmonar; I-D: izquierda-derecha; Qp:Qs: cociente de flujo pulmonar a sistémico; RVP: resistencia vascular pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo.
aAgrandamiento del VD con aumento del volumen de eyección.
bSiempre que no haya HAP o enfermedad del VI.
cPara los pacientes ancianos no aptos para cierre con dispositivo, se debe sopesar cuidadosamente el riesgo quirúrgico frente al potencial beneficio del cierre de la CIA.
dSopesar cuidadosamente el beneficio de eliminar el cortocircuito I-D frente el impacto potencialmente negativo del cierre de la CIA por aumento de la presión de llenado (considerando cierre, cierre fenestrado y no cerrar).
La reparación quirúrgica tiene una mortalidad baja (< 1% de los pacientes sin comorbilidad relevante), buenos resultados a largo plazo, esperanza de vida normal y baja morbilidad a largo plazo cuando se realiza pronto, en la niñez o la adolescencia, y en ausencia de HAP144,145. No obstante, la mortalidad quizá sea más alta en pacientes de edad avanzada o con comorbilidades.
Recomendaciones para la intervención en la comunicación interauricular (congénita y residual)
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Los pacientes con evidencia de sobrecarga de volumen del VDc y sin HAP (sin signos de aumento de la PAP no invasiva o confirmación invasiva de RVP < 3 UW en caso de tales signos) o enfermedad del VI deben someterse a cierre de la CIA con independencia de los síntomas146,147 | I | B |
| El cierre con dispositivo es el método de elección para el cierre de la CIA de tipo ostium secundum siempre que sea posible | I | C |
| Para los pacientes de edad avanzada no aptos para el cierre con dispositivo, se recomienda sopesar cuidadosamente el riesgo quirúrgico frente al beneficio potencial del cierre de la CIA | I | C |
| Es imprescindible la medición invasiva de la RVP de los pacientes que presenten signos de elevación de la PAP no invasiva | I | C |
| Para los pacientes con enfermedad del VI, se recomienda una prueba con balón y sopesar cuidadosamente el beneficio de eliminar el cortocircuito I-D frente al posible impacto negativo del cierre debido a un aumento en la presión de llenado (considerando cierre, cierre fenestrado y no cerrar) | I | C |
| Para los pacientes con sospecha de embolia paradójica (después de excluir otras causas), se debe considerar el cierre de la CIA con independencia del tamaño, siempre que no haya HAP ni enfermedad del VI | IIa | C |
| Para los pacientes con RVP 3-5 WU, se debe considerar el cierre de la CIA cuando haya un cortocircuito I-D relevante (Qp:Qs > 1,5) | IIa | C |
| Para los pacientes con RVP ≥ 5 WU, se puede considerar el cierre fenestrado de la CIA cuando la RVP descienda por debajo de 5 WU tras un tratamiento dirigido a la HAP y haya un cortocircuito I-D relevante (Qp:Qs > 1,5) | IIb | C |
| El cierre de la CIA debe evitarse en pacientes con fisiología de Eisenmenger o con HAP y RVP ≥ 5 WU a pesar del tratamiento dirigido a la HAP o cuando se produzca desaturación durante el ejerciciod | III | C |
CIA: comunicación interauricular; HAP: hipertensión arterial pulmonar; I-D: izquierda-derecha; PAP: presión arterial pulmonar; Qp:Qs: cociente de flujo pulmonar a sistémico; RVP: resistencia vascular pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cAgrandamiento del VD con aumento del volumen de eyección.
dHay pocos datos para establecer un punto de corte preciso pero, según la experiencia clínica, sería una reducción de la saturación arterial de oxígeno por debajo del 90%.
El cierre con dispositivo se ha convertido en la primera elección para el cierre del defecto del ostium secundum cuando es factible desde el punto de vista morfológico (diámetro forzado ≤ 38mm y borde suficiente de 5mm excepto hacia la aorta). Tal es el caso en un 80% de los pacientes. Aunque no se puede suponer que sea nula, varios estudios recientes no han constatado mortalidad alguna. Las complicaciones graves se observaron en menos del 1% de los pacientes148,149. Las taquiarritmias auriculares que ocurren poco después de la intervención son, en su mayoría, transitorias. La erosión de la pared auricular, la valva mitral anterior o la aorta y los eventos tromboembólicos parecen ser muy poco frecuentes150,151. El tratamiento antiagregante plaquetario es necesario durante al menos 6 meses con ácico acetilsalicílico 75 mg/día como mínimo. Aún se debe estudiar en mayor profundidad la incidencia potencial de las arritmias tardías o los efectos secundarios. Los estudios que comparan la cirugía con el cateterismo han constatado tasas de éxito y mortalidad similares, aunque con el cateterismo la morbilidad fue menor, las hospitalizaciones fueron más cortas y la tasa de reintervenciones, ligeramente mayor148,152.
El resultado es mejor si la reparación se realiza en pacientes de edad < 25 años144,145. Al parecer, el cierre de la CIA después de los 40 años no afecta a la frecuencia de aparición de arritmias durante el seguimiento146,153. No obstante, los pacientes se benefician del cierre a cualquier edad en lo que a morbilidad se refiere (capacidad de ejercicio, disnea, insuficiencia cardiaca derecha), sobre todo cuando puede realizarse por cateterismo146,153.
La mala función sistólica y diastólica del VI puede causar congestión pulmonar tras el cierre de la CIA y requerir una prueba antes de la intervención (oclusión con balón con revaluación hemodinámica) antes de decidir el tipo de tratamiento (cierre completo, fenestrado o no cerrar), teniendo en cuenta que un aumento de la presión de llenado secundario al cierre de la CIA puede empeorar los síntomas y el resultado clínico154.
Se debe evaluar con especial cuidado a los pacientes con HP. En estos casos, es imprescindible calcular la RVP. Si la RVP es < 5 UW, el cierre de la CIA es seguro, y está demostrado que tiene efectos beneficiosos en la PAP y los síntomas60,153,155. No obstante, incluso en este grupo, el grado de mejoría disminuye a medida que aumenta la PAP. Los pacientes con una RVP ≥ 5 UW tienen pocas posibilidades de mejoría60 e incluso pueden empeorar con el cierre completo de la CIA48,156. No se recomienda la prueba de vasorreactividad para tomar la decisión de cerrar una CIA en los pacientes con RVP ≥ 5 UW. Parece ser más seguro tratar la HAP, revaluar el estado hemodinámico durante el seguimiento y considerar un cierre fenestrado cuando la RVP caiga por debajo de 5 UW en presencia de un cortocircuito I-D significativo. Si no es así, se debe evitar el cierre de la CIA.
En pacientes con flutter auricular/FA, la crioablación o la ablación por radiofrecuencia (intervención en laberinto modificada) deberían plantearse en el momento de la cirugía. El cierre con dispositivo puede dificultar el acceso a la AI en futuras intervenciones electrofisiológicas.
Se debe sopesar cuidadosamente el riesgo quirúrgico individual de los pacientes de edad avanzada con CIA no apta para cierre con dispositivo frente a los beneficios potenciales del cierre de la CIA, por las comorbilidades.
4.1.5Aspectos específicos de la conexión venosa pulmonar anómalaLa conexión venosa pulmonar anómala puede aparecer asociada con CIA (típicamente en los defectos de los senos venosos) o aislada. Produce sobrecarga de volumen del VD, con un efecto fisiológico parecido al de la CIA, aunque en su forma aislada difiere en que no hay un cortocircuito D-I y la magnitud del cortocircuito I-D no está exacerbada por el desarrollo de una cardiopatía izquierda adquirida. La más común es la conexión de la vena pulmonar superior derecha a la VCI. Otras conexiones anómalas son la vena pulmonar derecha a la VCI (síndrome de la cimitarra, que puede asociarse con secuestro del lóbulo pulmonar inferior derecho), la vena pulmonar superior izquierda a la vena innominada izquierda y la vena pulmonar superior derecha conectada a la parte superior de la VCS. Las secuelas a largo plazo de las conexiones venosas pulmonares anómalas reflejan el impacto de la sobrecarga de volumen del VD y son similares a las secuelas de las CIA.
La reparación quirúrgica puede ser un reto, ya que el flujo venoso a baja velocidad conlleva un alto riesgo de trombosis de la vena operada, sobre todo en el síndrome de la cimitarra; solo la puede llevar a cabo un cirujano cardiaco especializado en CC.
Las indicaciones para la cirugía siguen los principios de recomendación para el cierre de la CIA, pero la idoneidad técnica para la reparación y el riesgo quirúrgico deben sopesarse frente al beneficio potencial de la intervención. No es frecuente que una única conexión venosa pulmonar anómala de un solo lóbulo pulmonar produzca una carga de volumen suficiente para justificar la reparación quirúrgica.
4.1.6Recomendaciones sobre el seguimientoLa evaluación de seguimiento debe incluir la determinación del cortocircuito residual, el tamaño y la función del VD, la IT y la PAP por ecocardiografía, así como la evaluación de las arritmias por antecedentes, ECG y, solo si está indicada, la monitorización Holter. Los pacientes menores de 25 años reparados sin secuelas ni lesiones residuales relevantes (sin cortocircuito residual, con PAP y VD normales y sin arritmias) no necesitan seguimiento regular. No obstante, tanto los pacientes como los médicos derivadores deberían conocer la posible aparición tardía de taquiarritmias.
Los pacientes con cortocircuito residual, PAP elevada o arritmias, antes o después de la reparación, y los reparados en la edad adulta, sobre todo mayores de 40 años, deben tener un seguimiento regular que incluya evaluación en centros especializados en CCA; los intervalos dependerán de la gravedad de las complicaciones residuales. Tras el cierre con dispositivo, se recomienda un seguimiento regular durante los primeros 2 años y luego cada 3–5 años, según los resultados.
Las arritmias posoperatorias tardías tras una reparación quirúrgica en pacientes menores de 40 años son, con mayor frecuencia, TRIA o flutter auricular y pueden tratarse con éxito mediante ablación por radiofrecuencia o crioablación. Con o sin reparación después de los 40 años, la FA se vuelve más común. En pacientes que se someten a cierre de la CIA después de los 40 años, la prevalencia de las arritmias auriculares es de un 40-60%. El acceso a la AI debe quedar restringido tras el cierre con dispositivo.
Puede aparecer estenosis de la VCS o de las venas pulmonares redirigidas después de la reparación de un defecto del seno venoso.
4.1.7Consideraciones adicionales–
Ejercicio/deporte: no hay restricciones para pacientes asintomáticos antes o después de la intervención que no tengan HP, arritmias relevantes o disfunción del VD; limitación a deportes recreativos de baja intensidad para pacientes con HAP (véase el apartado 3.5.5).
- •
Embarazo: el riesgo secundario al embarazo para las pacientes con HP es bajo, aunque puede haber riesgo de embolia paradójica. Se debe desaconsejar el embarazo a las pacientes con HP precapilar (véase el apartado 3.5.7).
- •
Profilaxis contra la EI: se recomienda para los 6 meses posteriores al cierre con dispositivo (véase el apartado 3.4.6).
En general, la comunicación interventricular (CIV) se diagnostica y, si está indicado, se trata antes de la edad adulta. Su cierre espontáneo es frecuente. Hay varias ubicaciones posibles del defecto dentro del septo interventricular, que pueden dividirse en 4 grupos; la nomenclatura varía, por lo que se añaden los sinónimos157:
- •
Perimembranoso, paramembranoso, subaórtico o conoventricular: es el más común, alrededor de un 80% de las CIV; localizado en el septo membranoso, con posible extensión al septo de entrada, trabecular o de salida; adyacente a las válvulas aórtica y tricúspide; los aneurismas del septo membranoso son frecuentes y pueden resultar en cierre parcial o completo.
- •
Muscular o trabecular: hasta un 15-20%; completamente rodeado de músculo; diferentes ubicaciones; frecuentemente múltiple; el cierre espontáneo es especialmente frecuente.
- •
Supracristal de salida, subarterial, subpulmonar, infundibular, supracristal, conal o yuxtaarterial de doble afección: aproximadamente un 5%; localizado debajo de las válvulas semilunares en el septo conal o de salida; a menudo se asocia con insuficiencia aórtica (IAo) progresiva por prolapso de las cúspides aórticas derechas, y aneurisma del seno de Valsalva.
- •
De entrada, canal AV o tipo de DSAV: entrada del septo ventricular inmediatamente inferior al aparato de la válvula AV; suele ocurrir en el síndrome de Down.
Con frecuencia hay un único defecto, aunque pueden ser múltiples. La CIV también es un elemento común de las anomalías complejas, como la TdF, la TGAcc, etc. El cierre espontáneo de la CIV puede ocurrir más a menudo en defectos musculares o trabeculares, pero también en perimembranosos. No es frecuente en los defectos de salida y ocurre principalmente durante la infancia158.
La dirección y la magnitud del cortocircuito están determinadas por la RVP y la resistencia vascular sistémica, el tamaño del defecto, la función sistólica y diastólica del VI o el VD y la presencia de obstrucción del tracto de salida del VD (OTSVD) y del VI (OTSVI).
4.2.2Presentación clínica e historia naturalLa presentación clínica habitual en adultos incluye:
- •
CIV intervenida en la infancia, sin CIV residual ni HP.
- •
CIV intervenida en la infancia, con CIV residual. El tamaño del cortocircuito residual determina la presencia de síntomas y el grado de sobrecarga de volumen del VI.
- •
CIV de pequeño tamaño con cortocircuito I-R irrelevante, sin sobrecarga de volumen del VI ni HP (CIV restrictiva), no se considera para la cirugía en la infancia.
- •
CIV con cortocircuito I-D, HP (diferentes grados) y varios grados de sobrecarga de volumen del VI; es poco común.
- •
Síndrome de Eisenmenger: CIV grande con cortocircuito D-I originalmente grande y desarrollo de enfermedad vascular pulmonar grave que resulta en inversión del cortocircuito y cianosis (véase los apartados 3.4.3 y 3.4.8).
La gran mayoría de los pacientes con CIV cerrada por completo en la infancia o con CIV pequeña nunca intervenidos o con defecto residual tras reparación quirúrgica sin sobrecarga de volumen del VI en ecocardiografía suelen permanecer asintomáticos y no necesitan cirugía159. No obstante, un porcentaje desconocido de pacientes con CIV residual pequeña sufren complicaciones con el paso del tiempo160. La supervivencia pasados los 40 años del cierre sigue siendo ligeramente inferior que la de la población general161.
Con el tiempo pueden producirse varias complicaciones:
- •
Se puede desarrollar VD con doble cámara (VDDC), que podría ser consecuencia de la lesión del endotelio del VD causada por un chorro de alta velocidad en la CIV.
- •
En el caso de una CIV de salida (supracristal), con menor frecuencia perimembranosa, hay un riesgo considerable de prolapso de la cúspide coronaria derecha o no coronaria de la válvula aórtica que resulte en IAo progresiva y formación de un aneurisma en el seno de Valsalva.
- •
Pueden aparecer arritmias, pero son menos frecuentes que en otras formas de CC162.
- •
El bloqueo cardiaco completo, hoy poco común, sí lo era en los primeros años de la cirugía cardiaca, por lo que puede darse sobre todo en pacientes mayores. Estos pacientes suelen necesitar marcapasos toda la vida.
- •
Disfunción tardía del VI e insuficiencia cardiaca.
- •
Endocarditis.
Véase el apartado 3.3 para conocer los principios generales.
Entre los hallazgos clínicos específicos, se encuentra el soplo holosistólico del tercer al cuarto espacio intercostal; se puede percibir una vibración precordial.
- •
La ecocardiografía es la técnica diagnóstica clave, ya que en general ofrece el diagnóstico y señala la gravedad de la enfermedad. Los hallazgos clave necesarios son ubicación, número y tamaño de los defectos, gravedad de la sobrecarga de volumen del VI y la PAP estimada. Debe evaluarse la IAo secundaria al prolapso de la cúspide derecha o no coronaria, sobre todo en caso de CIV de salida (supracristal) y perimembranosas altas. Deben excluirse el VDDC y el aneurisma en el seno de Valsalva.
- •
La RMC puede ser una alternativa si la ecocardiografía resulta insuficiente, sobre todo para evaluar la sobrecarga de volumen del VI y la cuantificación del cortocircuito.
- •
El cateterismo cardiaco es necesario en casos de PAP alta en ecocardiografía (PAP sistólica calculada > 40mmHg o signos indirectos cuando no pueda calcularse) para determinar la RVP.
- •
Los pacientes con HAP deben someterse a prueba de esfuerzo para excluir desaturación.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención de la comunicación interventricular (nativa y residual)» y la figura 3.
 DSV: defecto del septo ventricular; EI: endocarditis infecciosa;
DSV: defecto del septo ventricular; EI: endocarditis infecciosa; Tratamiento del defecto del septo ventricular. DSV: defecto del septo ventricular; EI: endocarditis infecciosa; HAP: hipertensión arterial pulmonar; Qp:Qs: cociente de flujo pulmonar a sistémico; IAo: insuficiencia aórtica; RVP: resistencia vascular pulmonar; VI: ventrículo izquierdo.
aAgrandamiento del VI con aumento del volumen de eyección.
bSe incluye a todos los pacientes con desaturación en reposo (fisiología de Eisenmenger) o ejercicio.
cSe requiere una decisión cuidadosa e individualizada en centros con experiencia.
El cierre quirúrgico puede realizarse con una mortalidad operatoria baja (1-2%) y buenos resultados a largo plazo163. Se puede considerar el cierre percutáneo para los pacientes con CIV residual o con muy mal acceso para el cierre quirúrgico y CIV musculares localizadas en el centro del septo interventricular. Se ha demostrado que es factible en CIV perimembranosas. Aún se desconoce si el riesgo de bloqueo AV completo y atrapamiento de la válvula tricúspide que causa la IAo o el riesgo de IAo observado en niños permanecen en los adultos.
Es infrecuente encontrar a pacientes que sean candidatos para el cierre de la CIV en la edad adulta. La mayoría de los pacientes tienen una CIV pequeña con cortocircuito irrelevante o ya se les ha desarrollado HP. Esto último debe evaluarse con especial cuidado, ya que los pacientes con cierre del cortocircuitio y HAP persistente o progresiva tienen un pronóstico particularmente malo48.
Recomendaciones para la intervención en el defecto del septo ventricular (congénito o residual)
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Los pacientes con sobrecarga de volumen del VIc sin HAP (ausencia de signos de aumento de la PAP no invasiva o confirmación invasiva de una RVP < 3 UW en caso de que los haya) deben someterse a cierre del DSV con independencia de los síntomas | I | C |
| Se debe consderar el cierre quirúrgico del DSV de los pacientes con antecedentes de EI incluso en ausencia de cortocircuito I-D relevante | IIa | C |
| Se debe considerar la cirugía para los pacientes con prolapso de una cúspide de la válvula aórtica secundario al DVS que causa IAo progresiva | IIa | C |
| Se debe considerar la cirugía para los pacientes que sufren HAP con RVP 3–5 UW si sigue habiendo cortocircuito I-D relevante (Qp:Qs > 1,5) | IIa | C |
| Los pacientes con HAP y RVP ≥ 5 UW podrían someterse a cierre del DSV si sigue habiendo cortocircuito I-D relevante (Qp:Qs > 1,5), aunque esta decisión debe ser individualizada y en centros especializados | IIb | C |
| El cierre del DSV debe evitarse en pacientes con fisiología de Eisenmenger o con HAP grave (RVP ≥ 5 UW) que sufran desaturación durante el ejerciciod | III | C |
DSV: defecto septal ventricular; EI: endocarditis infecciosa; HAP: hipertensión arterial pulmonar; IAo: insuficiencia aórtica; I-D: izquierdo-derecho; PAP: presión arterial pulmonar; Qp:Qs: cociente de flujo pulmonar a sistémico; RVP: resistencia vascular pulmonar; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cAgrandamiento del VI con aumento del volumen de eyección.
dHay pocos datos para establecer un punto de corte preciso, pero según la experiencia clínica, esto vendría dado por una caída de la saturación arterial de oxígeno por debajo del 90%.
Tanto el desarrollo de IAo o IT como el grado del cortocircuito (residual), la disfunción del VI, el aumento de la PAP y el desarrollo de VDDC deberían excluirse o evaluarse si están presentes en la ecocardiografía.
La posible aparición de bloqueo AV completo requiere atención; los pacientes que sufren bloqueo bifascicular o bloqueo trifascicular transitorio tras el cierre de la CIV tienen riesgo de bloqueo AV completo en los años siguientes.
Los pacientes con CIV no pequeñas, lesiones valvulares o deterioro hemodinámico (disfunción del VI o HAP) deben tener seguimiento anual con evaluación en centros especializados en CCA. En pacientes con CIV pequeña (nativo o residual, VI normal, PAP normal, asintomáticos) y sin ninguna otra lesión, el seguimiento en intervalos de 3-5 años es aceptable. Tras el cierre quirúrgico, se debe hacer seguimiento regular los primeros 2 años, dependiendo de los resultados, y posteriormente a intervalos de 2-5 años. Tras el cierre quirúrgico sin anomalía residual, los intervalos de 5 años son aceptables.
4.2.6Consideraciones adicionales- •
Ejercicio/deporte: no hay restricciones para pacientes tras el cierre de la CIV, o con CIV pequeña sin HP, arritmias relevantes ni disfunción del VI. Los pacientes con HAP deben limitarse a actividades/deportes recreativos de baja intensidad (véase el apartado 3.5.5).
- •
Embarazo: está contraindicado para las pacientes con HP precapilar (HAP). El riesgo es bajo para las pacientes asintomáticas con VI normal y sin HAP (véase el apartado 3.5.7).
- •
Profilaxis contra EI: se recomienda solo para los pacientes con alto riesgo (véase el apartado 3.4.6).
El DSAV (canal AV o defecto de la almohadilla endocárdica) se caracteriza por la presencia de un anillo AV común, con 5 valvas. En la forma parcial, las valvas puente anterior y posterior se funden en el centro y se crean orificios independientes a izquierda y derecha. En el DSAV completo, la fusión central no está y hay un único orificio. El DSAV parcial (DSA del ostium primum, canal AV parcial) tiene un defecto solo auricular. El DSAV completo (canal AV completo) tiene un defecto septal en la cruz cardiaca, que se extiende hasta los septos interauricular e interventricular. El nódulo AV está situado detrás y debajo del seno coronario. El haz de His y la rama izquierda quedan desplazados posteriormente. Esto representa una secuencia anómala de activación de los ventrículos (prolongación del tiempo de conducción AV, desviación del eje izquierdo) y es importante identificarlo en los estudios electrofisiológicos y la ablación con catéter.
La mayoría de los DSAV completos se producen en pacientes con síndrome de Down (> 75%) y la mayoría de los DSAV parciales ocurre en pacientes sin el síndrome (> 90%). Los DSAV pueden ocurrir junto con TdF u otras formas de CC compleja.
El DSAV con una posición desigual de la válvula AV común por encima de los ventrículos se acompaña de un grado variable de hipoplasia ventricular (DSAV desequilibrado). Las presentes recomendaciones se aplican a los DSAV equilibrados.
4.3.2Presentación clínica e historia naturalLa presentación clínica depende principalmente de la presencia y el tamaño del DSAV y la CIV y la capacidad de la válvula AV del lado izquierdo. Los síntomas no son específicos de un DSAV y son consecuencia del cortocircuito intracardiaco (I-D, D-I o bidireccional), la HP, la insuficiencia de la válvula AV, la disfunción ventricular o la OTSVI. Puede haber intolerancia al ejercicio, disnea, arritmia y cianosis. Podría haber una estenosis subaórtica (ESA) o aparecer con el tiempo. El bloqueo AV completo puede desarrollarse más tardíamente.
La historia del DSAV completo no intervenido es la del síndrome de Eisenmenger, a no ser que el DSV sea pequeño (véase los apartados 3.4.3 y 3.4.8).
El DSAV parcial no reparado es común en adultos. Los síntomas clínicos son los de un cortocircuito I-D auricular (véase el apartado 4.1) o insuficiencia de la válvula AV del lado izquierdo («fisura»). Los pacientes pueden estar asintomáticos, aunque los síntomas suelen incrementarse con la edad. A los 40 años, gran parte de los adultos están sintomáticos.
4.3.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos dependen de la variante individual (véase los apartados 4.3.1 y 4.3.2).
- •
La ecocardiografía es la técnica diagnóstica clave. Proporciona la evaluación de cada elemento anatómico del DSAV, las válvulas AV y sus conexiones (cabalgamiento), la gravedad y el sustrato exacto de la insuficiencia de la válvula AV, la magnitud y la dirección del cortocircuito intracardiaco, la función del VI y el VD, la PAP y la evaluación de la presencia/ausencia de ESA.
- •
La RMC está indicada cuando se requiere cuantificación adicional de los volúmenes ventriculares y su función, la insuficiencia de la válvula AV y la función del cortocircuito intracardiaco para tomar una decisión.
- •
El cateterismo cardiaco está indicado en caso de PAP alta en ecocardiografía (PAP sistólica calculada > 40mmHg o signos indirectos cuando no pueda calcularse) para evaluación de la RVP.
- •
Los pacientes con HAP deben realizar una prueba de esfuerzo para excluir la desaturación.
El cierre percutáneo del DSAV no es posible, por lo que la intervención es quirúrgica (cierre del defecto, reparación de la válvula). En casos de CIA o CIV residuales, el MP endocárdico supone alto riesgo de émbolos paradójicos. Esto debe tenerse en cuenta cuando esté indicado el MP. En ocasiones puede ser necesario un MP epicárdico. Se recomienda la experiencia de un cirujano cardiaco especialista en CC para todo tipo de cierres de DSAV y reparaciones de válvulas AV.
Recomendaciones para la intervención en el defecto del septo auriculoventricular
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| DSAV completo | ||
| Se debe evitar la reparación quirúrgica en pacientes con fisiología de Eisenmenger o con HAP grave (RVP ≥5 WU) que desarrollen desaturación durante el ejercicioc | III | C |
| Para la indicación de intervención, véase también DSV, apartado 4.2 | ||
| DSAV parcial (CIA delostiumprimum) | ||
| Un cirujano cardiaco experto en CC debe practicar el cierre quirúrgico en caso de sobrecarga de volumen del VD relevante | I | C |
| Para más detalles, véase CIA, apartado 4.1 | ||
| Insuficiencia de la válvula AV | ||
| Los pacientes sintomáticos con insuficiencia de la válvula AV moderada-grave deben someterse a cirugía valvular, preferiblemente reparación de la válvula AV, realizada por cirujanos cardiacos expertos en CC | I | C |
| Los pacientes asintomáticos con insuficiencia grave de la válvula del lado izquierdo y DTSVI ≥ 45mmd o FEVI ≤ 60% deben someterse a cirugía de la válvula tras haber excluido otras causas de disfunción del VI | I | C |
| En caso de FA o PAP sistólica > 50mmHg, debe considerarse la reparación quirúrgica para los pacientes asintomáticos con insuficiencia grave de la válvula AV del lado izquierdo, función del VI conservada (DTSVI < 45mmd o FEVI > 60%) y un sustrato de la insuficiencia con muchas probabilidades de resolverse con la reparación quirúrgica | IIa | C |
| Obstrucción del tracto de salida del ventrículo izquierdo | ||
| Véase el apartado 4.5.3 sobre intervención en ESA | ||
AV: auriculoventricular; CC: cardiopatías congénitas; CIA: comunicación interauricular; DSAV: defecto del septo auriculoventricular; DSV: defecto del septo ventricular; DTSVI: diámetro telesistólico del ventrículo izquierdo; ESA: estenosis subaórtica; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; HAP: hipertensión arterial pulmonar; OTSVI: obstrucción del tracto de salida del ventrículo izquierdo; PAP: presión arterial pulmonar; RVP: resistencia vascular pulmonar; VD: ventrículo derecho; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cHay pocos datos para establecer un punto de corte preciso, pero según la experiencia clínica, sería una reducción de la saturación arterial de oxígeno por debajo del 90%.
dEl punto de corte se refiere a adultos de tamaño medio y puede requerir adaptación en pacientes con estatura inusualmente corta o alta.
Se recomienda el seguimiento regular de por vida para todo paciente con DSAV, intervenido o no, que incluya evaluación en centros especializados en CCA. Debe prestarse especial atención al cortocircuito residual, la disfunción de la válvula AV, el agrandamiento y la disfunción del VI y el VD, la elevación de la PAP, las ESA y las arritmias164. La frecuencia de las visitas ambulatorias depende de la presencia y la gravedad de las anomalías residuales. Un DSAV reparado quirúrgicamente sin anomalías residuales relevantes debe tener seguimiento al menos cada 2-3 años. En el caso de anomalías residuales, los intervalos deben ser más cortos.
Las indicaciones para la reintervención por anomalías residuales son comparables a las de la cirugía primaria. En pacientes intervenidos, el problema más frecuente es la insuficiencia de la válvula AV del lado izquierdo165,166. La estenosis de la válvula AV del lado izquierdo sintomática, en la mayoría de los casos consecuencia de una reparación anterior, debe intervenirse.
4.3.6Consideraciones adicionales–
- •
Ejercicio/deporte: para la mayoría de los pacientes con DSAV reparado y sin complicaciones, la actividad física no requiere restricciones. No obstante, muchos tendrán un rendimiento por debajo de lo normal medido objetivamente. Los pacientes con complicaciones residuales importantes requieren recomendaciones individuales (véase el apartado 3.5.5).
- •
Embarazo: lo toleran bien las pacientes con reparación completa sin lesiones residuales relevantes. Un DSAV parcial no intervenido supone un riesgo mayor de embolización paradójica. El embarazo está contraindicado para las pacientes con HP precapilar. Como norma, las pacientes con insuficiencia de la válvula AV del lado izquierdo que no tienen indicación para cirugía toleran el embarazo relativamente bien, aunque pueden sufrir arritmias y empeoramiento de la insuficiencia167 (véase el apartado 3.5.7).
- •
Profilaxis contra la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
El DAP consiste en la comunicación persistente entre la AP izquierda y la aorta descendente distal a la arteria subclavia izquierda. Puede estar asociado a una variedad de lesiones de CC. No obstante, suele tratarse de un hallazgo aislado en el adulto.
En un principio, el DAP resulta en cortocircuito I-D y sobrecarga de volumen del VI y la AI. En DAP moderados y grandes, la PAP aumenta. En pacientes que llegan a la edad adulta con un DAP moderado, puede que la sobrecarga de volumen del VI o la HAP sean predominantes. Los pacientes adultos con DAP en general han adquirido fisiología de Eisenmenger.
4.4.2Presentación clínica e historia naturalLa presentación en los pacientes adultos con DAP incluye:
- •
Ductus pequeño sin sobrecarga de volumen del VI (VI normal) y PAP normal, generalmente asintomática.
- •
DAP moderado con sobrecarga predominante de volumen del VI: VI grande con función normal o reducida, con posible insuficiencia cardiaca izquierda.
- •
DAP moderado con HAP predominante: sobrecarga de presión del VD, con posible insuficiencia cardiaca derecha.
- •
DAP grande: fisiología de Eisenmenger con hipoxemia diferencial y cianosis diferencial, extremidades inferiores cianóticas, a veces el brazo izquierdo también (véase las secciones 3.4.3 y 3.4.8).
La formación de aneurismas del ductus es una complicación poco común.
4.4.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos específicos incluyen un soplo continuo que desaparece con el desarrollo del síndrome de Eisenmenger (para la cianosis diferencial, véase el apartado 4.4.2). La saturación de oxígeno debe medirse en las extremidades superiores e inferiores.
- •
La ecocardiografía es la técnica diagnóstica clave, ya que proporciona el diagnóstico (aunque quizá sea difícil en pacientes con fisiología de Eisenmenger), el grado de sobrecarga de volumen del VI, la PAP, el tamaño de la AP y los cambios del corazón derecho.
- •
La RMC está indicada cuando se necesita cuantificación adicional de los volúmenes del VI y cuantificar el cortocircuito (Qp:Qs).
- •
La TCC o la RMC permiten la evaluación de la anatomía de la AP cuando sea necesario.
- •
El cateterismo cardiaco está indicado en el caso de PAP alta en ecocardiografía (PAP sistólica calculada > 40mmHg o signos indirectos cuando no pueda calcularse) para evaluación de la RVP. La medición del flujo sanguíneo pulmonar puede ser difícil en este contexto. Es fundamental medir la saturación de oxígeno en las AP izquierda y derecha.
- •
Los pacientes con HAP deben pasar una prueba de esfuerzo para excluir la desaturación en las extremidades inferiores.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención en el ductus arterioso persistente» y la figura 4.
 DAP: ductus arterioso persistente;
DAP: ductus arterioso persistente; Tratamiento del ductus arterioso persistente. DAP: ductus arterioso persistente; HAP: hipertensión arterial pulmonar; Qp:Qs: cociente de flujo pulmonar a sistémico; RVP: resistencia vascular pulmonar; VI: ventrículo izquierdo.
aAgrandamiento del VI con aumento del volumen de eyección.
bSe incluye a todos los pacientes con desaturación de las extremidades inferiores en reposo (fisiología de Eisenmenger) o ejercicio.
cSe requiere una decisión cuidadosa e individualizada en centros con experiencia.
En adultos, la calcificación del DAP podría ser un problema para el cierre quirúrgico. El cierre con dispositivo es el método de elección, incluso si las intervenciones cardiacas están indicadas por otras lesiones cardiacas concomitantes, y se puede realizar con éxito en la gran mayoría de los adultos, con una tasa de complicaciones muy baja168–170. La cirugía se reserva para los pacientes poco comunes con un ductus demasiado largo para el cierre con dispositivo o con anatomía inadecuada, como la formación de aneurismas.
Recomendaciones para la intervención en el ductus arterioso persistente
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Se debe cerrar el DAP de los pacientes con sobrecarga de volumen del VIc sin HAP (ausencia de signos de aumento de la PAP no invasiva o confirmación invasiva de RVP < 3 UW en caso de que los haya) con independencia de los síntomas | I | C |
| Se recomienda el cierre con dispositivo como el método de elección cuando sea técnicamente factible | I | C |
| Se debe considerar la cirugía para los pacientes que sufren HAP con RVP 3-5 UW si sigue habiendo cortocircuito I-D relevante (Qp:Qs > 1,5) | IIa | C |
| Los pacientes que sufren HAP con RVP ≥ 5 UW podrían someterse a cierre del DAP si sigue habiendo cortocircuito I-D relevante (Qp:Qs > 1,5), aunque esta decisión debe ser individualizada y en centros especializados | IIb | C |
| Se debe evitar el cierre del DAP de pacientes con fisiología de Eisenmenger o que sufran desaturación en las extremidades inferiores durante el ejerciciod | III | C |
DAP: ductus arterioso persistente; HAP: hipertensión arterial pulmonar; I-D: izquierda-derecha; PAP: presión arterial pulmonar; Qp:Qs: cociente de flujo pulmonar a sistémico; RVP: resistencia vascular pulmonar; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cAgrandamiento del VI con aumento del volumen de eyección.
dHay pocos datos para establecer un punto de corte preciso, pero según la experiencia clínica, sería una disminución de la saturación arterial de oxígeno por debajo del 90%.
La evaluación ecocardiográfica debe incluir tamaño y función del VI, PAP, cortocircuito residual y lesiones asociadas.
Los pacientes sin cortocircuito residual, VI normal y PAP normal no requieren seguimiento regular después de los 6 meses.
Los pacientes con disfunción del VI o con HAP residual deben tener seguimiento en intervalos de 1-3 años, según la gravedad, y con evaluación en centros especializados en CCA.
4.4.6Consideraciones adicionales–
- •
Ejercicio/deporte: no hay restricciones para pacientes asintomáticos ni antes ni después de la intervención siempre que no tengan HP; limitación a deportes de baja intensidad para pacientes con HAP.
- •
Embarazo: no hay riesgo significativamente aumentado consecuencia del embarazo para las pacientes sin HP. El embarazo está contraindicado para las pacientes con HP precapilar (véase el apartado 3.5.7).
- •
Profilaxis contra la EI: limitada a pacientes en alto riesgo (véase el apartado 3.4.6).
La causa más común de estenosis de la válvula aórtica congénita es la VAB. En hasta un 80% de los pacientes con VAB va a desarrollarse dilatación progresiva de la aorta ascendente, que se trata en el apartado 4.7.2. Para el tratamiento de la IAo secundaria a VAB, véase la guía de la ESC/EACTS de 2017 sobre el tratamiento de la cardiopatía valvular25.
4.5.1.2Presentación clínica e historia naturalLos pacientes suelen permanecer asintomáticos durante muchos años. El avance de la estenosis varía y depende de la gravedad inicial, el grado de calcificación, la edad y factores de riesgo de ateroesclerosis. En la VAB, el avance es más rápido en los pacientes con mayor excentricidad de la línea de cierre o línea de cierre orientada ateroposteriormente.
El pronóstico es bueno y la muerte súbita es poco común en pacientes asintomáticos, que toleran bien el ejercicio incluso cuando la estenosis es grave171.
Una vez aparecen los síntomas (angina de pecho, disnea o síncope), el pronóstico se deteriora rápidamente. En pacientes con VAB, la mortalidad cardiaca se ha constatado en un 0,3% por paciente-año de seguimiento; la frecuencia de la disección aórtica, en un 0,03% y la endocarditis, en un 0,3%. Se ha descubierto que los senos aórticos o la aorta ascendente están dilatados en el 45% de los pacientes a los 9 años de seguimiento172.
4.5.1.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales. Los criterios diagnósticos para la clasificación de la gravedad de la EA se resumen en la tabla 12.
Criterios diagnósticos del grado de estenosis aórtica173
| EA leve | EA moderada | EA grave | |
|---|---|---|---|
| Vmáx (m/s)* | 2,6-2,9 | 3,0-3,9 | ≥ 4,0 |
| Gradiente medio (mmHg)a | < 20 | 20-39 | ≥ 40 |
| AVA (cm2) | > 1,5 | 1,0-1,5 | < 1,0 |
| AVAi (cm2/m2 de ASC) | > 0,85 | 0,60-0,85 | < 0,60 |
| Velocidad del TSVI/velocidad de la válvula aórtica | > 0,50 | 0,25-0,50 | < 0,25 |
ASC: área de superficie corporal; AVA: área valvular aórtica; AVAi: área valvular aórtica indexada; EA: estenosis aórtica; TSVI: tracto de salida del ventrículo izquierdo; Vmáx: velocidad Doppler máxima.
*Con flujo transvalvular normal.
Los hallazgos clínicos específicos incluyen el típico soplo sistólico de eyección sobre la válvula aórtica hacia las arterias carótidas. Puede notarse una vibración. El ECG puede revelar hipertrofia ventricular izquierda (HVI) con o sin distensión. Debe excluirse la CoA en los pacientes con diagnóstico de VAB (véase el apartado 4.6).
- •
La ecocardiografía es la técnica de referencia para el diagnóstico de la EA y la evaluación del grado de calcificación, la función del VI, la HVI y las lesiones asociadas, incluida la dilatación de la aorta ascendente. Con la ecocardiografía Doppler se determina la gravedad de la EA a partir de la velocidad transvalvular máxima (Vmáx), el gradiente medio y el área valvular aórtica (AVA) calculada por la ecuación de continuidad. Para más detalles, véase las recientes recomendaciones para la evaluación de la EA173.
- •
La ETE a veces puede ser útil para planimetrar el AVA en válvulas no calcificadas.
- •
Se recomiendan las pruebas de ejercicio para los pacientes asintomáticos, especialmente con EA de moderada a grave, para confirmar el estado asintomático y evaluar la tolerancia al ejercicio, la respuesta de la presión arterial, las arritmias para la estratificación del riesgo y el momento de la cirugía.
- •
La ecocardiografía con dobutamina en dosis bajas es útil en la EA con función del VI reducida (flujo bajo, gradiente de EA bajo)173.
- •
La RMC o la TC, a pesar de su potencial para evaluar la EA, son necesarias principalmente para valorar la dilatación de la aorta ascendente, que quizá sea indetectable en la ecocardiografía cuando ocurre distal a la unión senotubular.
- •
La TCC se ha convertido en una herramienta particularmente útil para cuantificar la calcificación valvular al valorar la gravedad en la EA de gradiente bajo, aunque es importante tener en cuenta que en pacientes jóvenes la estenosis no se asocia necesariamente con calcificación significativa.
- •
El cateterismo cardiaco solo es necesario cuando la evaluación no invasiva aporta resultados dudosos y para exminar las arterias coronarias o la posibilidad de angioplastia.
Recomendaciones para la intervención en la estenosis aórtica
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Los pacientes con síntomas espontáneos o en la prueba de ejercicio y gradiente Doppler medio ≥ 40mmHg deben someterse a cirugía | I | C |
| Los pacientes con gradiente Doppler medio < 40mmHg deben someterse a cirugía cuando tengan: | I | C |
| • Síntomas atribuibles a una obstrucción (disnea de esfuerzo, angina, síncope) | ||
| • Disfunción sistólica del VI (FE < 50%) sin otra explicación | ||
| • Si requieren cirugía por EC o valvulopatía relevantes | ||
| Se podría considerar la reparación para los pacientes con gradiente Doppler medio ≥ 40mmHgc, pero sin síntomas, disfunción sistólica del VI, HVI ni prueba de ejercicio anómala si el riesgo quirúrgico es bajo | IIb | C |
BNP: péptido natriurético cerebral; CABG: cirugía de revascularización coronaria; EA: estenosis aórtica; FE: fracción de eyección; FEVI: fracción de eyección del VI; HP: hipertensión pulmonar; PAP: presión arterial pulmonar; VI: ventrículo izquierdo; Vmáx: velocidad máxima por Doppler.
aClase de recomendación.
bNivel de evidencia.
Los pacientes sintomáticos requieren cirugía urgente. El tratamiento médico para la insuficiencia cardiaca se reserva únicamente para pacientes inoperables. Hasta el momento, no se ha demostrado que el tratamiento con estatinas u otro tratamiento médico retrasen el avance de la EA.
4.5.1.5Tratamiento quirúrgico/cateterismo intervencionistaLas indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención en la estenosis valvular aórtica» y la figura 5.
 ESA: estenosis subaórtica;
ESA: estenosis subaórtica; Tratamiento de la obstrucción grave del tracto de salida del ventrículo izquierdo. EA: estenosis aórtica; EAsupra: estenosis aórtica supravalvular; ESA: estenosis subaórtica; FEVI: fracción de eyección del ventrículo izquierdo; HP: hipertensión pulmonar; OTSVI: obstrucción del tracto de salida del ventrículo izquierdo; PAP: presión arterial pulmonar.
aVéase el apartado 4.5. Existen diferencias fundamentales en las decisiones de tratamiento en comparación con la EA valvular, sobre todo porque no suele ser necesaria la sustitución de la válvula.
bVelocidad pico > 5,5 m/s; calcificación grave + progresíón de la velocidad pico ≥ 0,3 m/s/año; elevación relevante de neurohormonas (> 3 veces el intervalo normal corregido por edad y sexo); HP grave (PAP sistólica > 60mmHg sin otra explicación).
Para adolescentes y adultos jóvenes con válvulas no calcificadas seleccionados, puede considerarse la valvuloplastia con balón bien como tratamiento puente para pacientes hemodinámicamente inestables, bien para retrasar la sustitución valvular a mujeres con adecuada anatomía valvular que desean gestar. En pacientes con válvulas calcificadas, el tratamiento de elección es la sustitución de la válvula. Las válvulas mecánicas son más duraderas que las biológicas o los homoinjertos, pero requieren tratamiento anticoagulante de por vida. Se ha propuesto la intervención de Ross para las pacientes en edad de tener hijos y para quienes quieren evitar la anticoagulación. Se trata de una intervención de mayor envergadura (operación de 2 válvulas) con una significativa tasa reintervenciones por la degeneración progresiva tras el homoinjerto. El implante percutáneo de la válvula se ha convertido en una técnica alternativa para tratar la estenosis del sustituto de la válvula pulmonar (el homoinjerto). En la actualidad, el implante percutáneo de la válvula aórtica no tiene lugar en el tratamiento contra la EA congénita, salvo en raras excepciones de riesgo quirúrgico alto.
4.5.1.6Recomendaciones sobre el seguimientoSe requiere un seguimiento regular de por vida a intervalos que dependen de la gravedad de la estenosis. También es necesario a intervalos anuales tras intervención de la válvula.
Es obligatorio tomar imágenes ecocardiográficas de la válvula y la raíz aórtica para determinar el avance de la estenosis valvular y la dilatación aórtica. Si la aorta ascendente no puede visualizarse bien en la ETT o el diámetro de la raíz es > 40mm, se recomiendan la RMC o la TCC de la aorta de los pacientes con VAB o antecedente de sustitución valvular aislada176.
4.5.1.7Consideraciones adicionales- •
Ejercicio/deporte: los pacientes con EA sintomática grave y asintomática, así como aquellos con estenosis moderada secundaria a VAB y aorta dilatada, deben evitar la práctica de ejercicio intenso o isométrico y deportes de competición. En la EA de leve a moderada, el deporte sí está permitido. Se recomienda realizar antes una prueba de ejercicio para el asesoramiento24.
- •
Embarazo: está contraindicado en la EA sintomática grave. El tratamiento por valvulotomía con balón o cirugía debe realizarse antes de la concepción. Para las pacientes asintomáticas con EA grave y una prueba de ejercicio normal seleccionadas, el embarazo sería posible, aunque hay controversia. La aorta requiere especial atención, ya que la dilatación aórtica secundaria a la VAB puede haberse producido y avanzar durante el embarazo. Hay riesgo de disección (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
La EA supravalvular (EASV) puede ocurrir como parte del síndrome de Williams-Beuren o aparecer de forma aislada/familiar, causada respectivamente por una deleción del gen de la elastina en el cromosoma 7q11.23 con pérdida de función o por una mutación en ese mismo gen. Causa arteriopatía obstructiva de diversa gravedad, que es más pronunciada en la unión sinotubular177.
La EASV también puede encontrarse en el contexto de la hipercolesterolemia familiar homocigótica178. Puede presentarse como un diafragma fibroso localizado distal al ostium de la arteria coronaria o, más frecuentemente, como una deformidad externa en reloj de arena con el correspondiente estrechamiento luminal de la aorta, o como estenosis difusa de la aorta ascendente. Suele ocurrir como parte del síndrome de Williams-Beuren, y puede deberse a hipoplasia de toda la aorta, afección del ostium coronario o estenosis de ramas principales de la aorta o las arterias pulmonares.
4.5.2.2Presentación clínica e historia naturalLa mayoría de los pacientes muestran síntomas de obstrucción del tracto de salida o isquemia miocárdica durante la infancia. Aunque la EASV no suele progresar en la edad adulta, el riesgo de complicaciones cardiacas permanece179. La muerte súbita es muy poco frecuente, pero es más común en la EASV con síndrome de Williams-Beuren, estenosis periférica difusa de la AP o EC, sobre todo durante los procedimientos anestésicos.
4.5.2.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
La auscultación revela un fuerte soplo sistólico de eyección que se aprecia mejor en el borde esternal inferior izquierdo y sin soplo diastólico de IAo acompañante.
- •
La ecocardiografía permite el diagnóstico anatómico de la EASV. La ecocardiografía Doppler proporciona gradientes de presión, aunque se puede sobrestimar la verdadera reducción de la presión de la obstrucción. La ETE permite una buena visualización del ostium coronario; la ETE 3D se puede usar para la definición anatómica precisa de la región estenosada180.
- •
Para la prueba de ejercicio, véase EA valvular en el apartado 4.5.1.
- •
La RMC y la TCC proporcionan una definición anatómica precisa de la lesión e identifican lesiones adicionales en la aorta y sus ramas (estenosis de las arterias carótida y renal) y las arterias coronarias y pulmonares.
- •
El cateterismo cardiaco: se recomienda la valoración hemodinámica cuando la cuantificación no invasiva resulte dudosa.
- •
Resulta útil la evaluación genética con asesoramiento y pruebas mediante técnicas de micro-array para el diagnóstico del síndrome de Williams-Beuren, y la secuenciación del gen de la elastina en las presentaciones no sindrómicas.
La cirugía es el tratamiento primario. La tasa de mortalidad operatoria del diafragma fibroso y la deformidad en reloj de arena es < 5%. Puesto que las arterias coronarias están a una elevada presión, la cirugía podría considerarse más precozmente que para los pacientes con estenosis valvular aórtica, especialmente cuando no se requiere sustituir la válvula. Después de la reparación operatoria, la tasa de supervivencia es de un 80-85% a los 20 años181. Puede hallarse IAo en un 25% de los pacientes, pero no suele ser progresiva tras el alivio quirúrgico de la EASV.
Recomendaciones para la intervención en la estenosis aórtica supravalvular
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Los pacientes con síntomas espontáneos o en la prueba de ejercicio y gradiente Doppler medio ≥ 40mmHg deben someterse a cirugía | I | C |
| Los pacientes con gradiente Doppler medio < 40mmHg deben someterse a cirugía cuando tengan: | I | C |
| • Síntomas atribuibles a una obstrucción (disnea de esfuerzo, angina, síncope) | ||
| • Disfunción sistólica del VI (FE < 50%) sin otra explicación | ||
| • Si requieren cirugía por EC o valvulopatía relevantes | ||
| Se podría considerar la reparación para los pacientes con gradiente Doppler medio ≥ 40mmHgc, pero sin síntomas, disfunción sistólica del VI, HVI ni prueba de ejercicio anómala si el riesgo quirúrgico es bajo | IIb | C |
EC: enfermedad coronaria; FE: fracción de eyección; HVI: hipertrofia ventricular izquierda; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cLos gradientes derivados del Doppler quizá sobrestimen la obstrucción y haya que confirmarlos por cateterización cardiaca izquierda.
Es necesario un seguimiento regular de por vida, con ecocardiografía para determinar el avance de la obstrucción (poco frecuente), el tamaño y la función del VI y la aparición de síntomas, así como tras la cirugía para detectar la reestenosis tardía, el desarrollo de aneurismas (RMC/TCC) y la aparición o avance de EC. El seguimiento debería incluir evaluación en centros especializados en CCA.
4.5.2.6Consideraciones adicionales- •
Ejercicio/deporte: véase estenosis valvular aórtica en el apartado 4.5.1.
- •
Embarazo: véase estenosis valvular aórtica en el apartado 4.5.1. Los pacientes con síndrome de Williams-Beuren y mutaciones en el gen de la elastina tienen un riesgo de transmisión del 50%, por lo que se recomienda el cribado familiar.
- •
Profilaxis de la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
La ESA puede ocurrir como lesión única, aunque suele estar relacionada con DSV, DSAV o complejo de Shone o puede aparecer tras la corrección de estas lesiones. Es consecuencia de una línea fibrosa en el tracto de salida del ventrículo izquierdo (TSVI) proximal a la válvula aórtica o como estrechamiento fibromuscular y debe diferenciarse de la cardiomiopatía hipertrófica.
4.5.3.2Presentación clínica e historia naturalEl curso clínico es muy variable. La presencia de CC, especialmente DSV, está relacionada con la progresión de la ESA; no parece que la edad tenga papel alguno. La IAo es frecuente, pero rara vez tiene importancia o progresión hemodinámica182. Aunque es muy poco frecuente, se ha documentado muerte súbita de pacientes con ESA.
4.5.3.3Estudio diagnósticoVéase el apartado 3.3. para conocer los principios generales.
Los hallazgos clínicos incluyen un soplo sistólico de eyección en el borde esternal izquierdo y el vértice sin radiación a las carótidas y sin clic sistólico de eyección. Un soplo diastólico indica IAo.
- •
La ecocardiografía visibiliza la anatomía del TSVI, las anomalías de la válvula aórtica concomitantes, el grado de IAo, la función del VI, la HVI y las lesiones relacionadas. Con la ecocardiografía Doppler se determina la gravedad de la obstrucción subvalvular, pero los gradientes derivados del Doppler pueden sobrestimar la obstrucción y habría que confirmarla por cateterismo cardiaco. A veces, la ETE es necesaria para demostrar la membrana. La ETE 3D puede ser útil para caracterizar la compleja anatomía del TSVI y planimetrar el área de obstrucción.
- •
La RMC puede ser útil para caracterizar la compleja anatomía del TSVI, sobre todo en pacientes con mala ventana acústica.
El tratamiento quirúrgico es la única intervención eficaz e implica la resección circunferencial del anillo fibroso y partes de la base muscular a lo largo de la superficie septal izquierda. La ESA fibromuscular o tipo túnel requiere una resección más extensa o una intervención de Konno. Los resultados quirúrgicos son buenos, pero puede haber reestenosis. En pacientes con riesgo quirúrgico bajo y morfología apropiada para la reparación, el umbral para la intervención es más bajo que en la EA, sobre todo porque el implante no es necesario. En el caso de IAo moderada o grave, se debe sustituir la válvula aórtica en el momento de la cirugía.
4.5.3.5Recomendaciones sobre el seguimientoPara los no operados, es necesario un seguimiento regular de por vida con ecocardiografía para determinar el avance de la obstrucción, la IAo y la función y el tamaño del VI. También es necesario un seguimiento posoperatorio regular para detectar reestenosis tardía (frecuente, sobre todo en formas aisladas y con tratamiento quirúrgico en la infancia), IAo progresiva, complicaciones como arritmias, bloqueo cardiaco y DSV iatrogénico. El seguimiento debe incluir evaluación en centros especializados en CCA, con una frecuencia que dependerá de la progresión esperada de la enfermedad.
Recomendaciones para la intervención en la estenosis subaórtica
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Los pacientes con síntomas espontáneos o en la prueba de ejercicio y un gradiente Doppler medio ≥ 40mmHgc o IAo grave deben someterse a cirugía | I | C |
| Se debe considerar la cirugía para los pacientes asintomáticos si: | IIa | C |
| • La FEVI es < 50% (el gradiente puede ser < 40 mmHg) o | ||
| • La IAo es grave y el DTSVI es > 50mm (o 25 mm/m2 de ASC) o la FE es < 50%d | ||
| • El gradiente Doppler medio es ≥ 40mmHgc y hay HVI significativa | ||
| • El gradiente Doppler medio es ≥ 40mmHgc y la respuesta de la presión arterial en la prueba de ejercicio es anómala | ||
| Se puede considerar la cirugía para los pacientes asintomáticos: | IIb | C |
| • Si el gradiente Doppler medio es ≥ 40mmHgc, el VI es normal (FE > 50% y ausencia de HVI), la prueba de ejercicio es normal y el riesgo quirúrgico es bajo | ||
| • Si la progresión de la IAo está documentada y se vuelve más leve, para prevenir una futura progresión | ||
ASC: área de superficie corporal; DTSVI: diámetro telesistólico del ventrículo izquierdo; ESC: Sociedad Europea de Cardiología; FE: fracción de eyección; FEVI: fracción de eyección del VI; HVI: hipertrofia ventricular izquierda; IAo: insuficiencia aórtica; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
cLos gradientes derivados del Doppler quizá sobrestimen la obstrucción y haya que confirmarlos por cateterización cardiaca izquierda.
dVéase la guía de la ESC de 2017 sobre el tratamiento de las valvulopatías25.
- •
Ejercicio/deporte: véase estenosis valvular aórtica en el apartado 4.5.1.
- •
Embarazo: solo está contraindicado en la ESA sintomática grave, en cuyo caso la cirugía debe realizarse antes del embarazo; debe considerarse incluso en caso de ESA asintomática grave (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
Se considera que la CoA es parte de una arteriopatía generalizada, y no solo un estrechamiento circunscrito a la aorta. Ocurre como estenosis discreta o como un segmento aórtico largo e hipoplásico. Normalmente, la CoA se localiza en el área donde se inserta el ductus arterioso, y es ectópica en muy pocos casos (aorta ascendente, descendente o abdominal).
Las lesiones vinculadas incluyen VAB, hasta el 85%, aneurisma de la aorta ascendente, EA subvalvular, valvular o supravalvular, estenosis de la válvula mitral (válvula mitral en paracaídas, un complejo conocido como síndrome de Shone) o defectos cardiacos congénitos complejos. También puede estar relacionada con los síndromes de Turner y Williams-Beuren. Se ha documentado la presencia de anomalías vasculares extracardiacas, como un origen anómalo de la subclavia derecha en un 4-5% de los casos y circulación arterial colateral y aneurismas intracerebrales en hasta el 10%.
4.6.2Presentación clínica e historia naturalLos signos y síntomas dependen de la gravedad de la CoA. Los pacientes con CoA grave ya tienen signos y síntomas en etapas tempranas de la vida, mientras los casos especialmente leves pueden no manifestarse hasta la edad adulta, cuando se detecta al tratar la HTA.
Los síntomas clave pueden incluir cefalea, hemorragia nasal, mareos, tinnitus, disnea, angina abdominal, claudicación, calambres en las piernas, fatiga en las piernas por esfuerzo y pies fríos.
Los pacientes con CoA que alcanzan la adolescencia tienen una muy buena supervivencia hasta los 60 años. La morbilidad a largo plazo es frecuente, y se debe fundamentalmente a las complicaciones aórticas y la hipertensión183.
El curso natural podría verse complicado por insuficiencia cardiaca izquierda, hemorragia intracraneal (por aneurisma en forma de baya), EI, rotura o disección aórtica, enfermedad prematura de las arterias coronarias y cerebrales y defectos cardiacos asociados.
4.6.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
La medición de la presión arterial en consulta en las extremidades inferiores y superiores es el estudio primario de todos los pacientes con CoA. Un gradiente de presión arterial sistólica ≥ 20mmHg entre las extremidades superiores y las inferiores indica una CoA relevante. El pulso débil o ausente en las extremidades inferiores y el retraso del pulso radial-femoral también indica CoA relevante.
- •
Se recomienda la determinación ambulatoria de la presión arterial en el brazo derecho para detectar/confirmar la hipertensión arterial (presión sistólica media de 24 h > 130mmHg o diastólica > 80mmHg).
- •
Otros hallazgos son frémito supraesternal (obstrucción proximal), soplo interescapular (sistólico) o soplos continuos (por los vasos colaterales). En caso de CoA puntual, los soplos pueden estar completamente ausentes.
- •
Los hallazgos de la radiografía torácica quizá incluyan muescas costales de la tercera y la cuarta (hasta la octava) costilla desde las colaterales.
- •
La ecocardiografía proporciona información sobre el punto, la estructura y el grado de CoA, la función y la hipertrofia del VI, las anomalías cardiacas asociadas y los diámetros de los vasos aórticos y supraaórticos. Los gradientes del Doppler no son útiles para la cuantificación en la coartación nativa ni en la posoperatoria. En presencia de colaterales extensas, los gradientes no son fiables y suelen subestimarse. Tras la reparación quirúrgica o el implante de un stent, pueden producirse mayores índices de flujo sistólico, incluso en ausencia de estrechamiento relevante, por una falta de adaptabilidad aórtica y recuperación de la presión después del Doppler. En estos casos, el gradiente está sobrestimado. Se supone que un fenómeno diastólico de «fuga» es el signo más fiable de coartación o recoartación imortantes.
- •
La RMC y la TCC, con reconstrucción 3D, son las técnicas no invasivas de elección para evaluar toda la aorta en adultos. Ambas representan el punto, el alcance y el grado de estrechamiento de la aorta, el arco aórtico, la aorta preestenótica y posestenótica y las colaterales. Ambos métodos detectan complicaciones como aneurismas, reestenosis o estenosis residual184.
- •
Está indicada la imagen de los vasos intracerebrales en caso de síntomas o manifestaciones clínicas de aneurisma/rotura.
- •
El cateterismo cardiaco con manometría (un gradiente pico-pico ≥20mmHg indica una CoA hemodinámicamente significativa en ausencia de colaterales bien desarrolladas) y se realiza en el contexto del tratamiento intervencionista. Es importante tener en cuenta que la anestesia general durante la determinación invasiva puede subestimar el gradiente.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención de la coartación y la recoartación aórtica» y la figura 6.
 CoA: coartación aórtica. aSe debe considerar la monitorización ambulatoria de la presión arterial del brazo derecho para el diagnóstico de hipertensión. bMedición confirmada de forma invasiva. cRelativa al diámetro aórtico a nivel del diafragma.' title='Tratamiento de la coartación y recoartación aórtica.
CoA: coartación aórtica. aSe debe considerar la monitorización ambulatoria de la presión arterial del brazo derecho para el diagnóstico de hipertensión. bMedición confirmada de forma invasiva. cRelativa al diámetro aórtico a nivel del diafragma.' title='Tratamiento de la coartación y recoartación aórtica. Tratamiento de la coartación y recoartación aórtica. CoA: coartación aórtica.
aSe debe considerar la monitorización ambulatoria de la presión arterial del brazo derecho para el diagnóstico de hipertensión.
bMedición confirmada de forma invasiva.
cRelativa al diámetro aórtico a nivel del diafragma.
En la CoA nativa y la recoartación con anatomía apropiada, el procedimiento con stent se ha convertido en el tratamiento de primera elección para los adultos en muchos centros185. Son preferibles los stents recubiertos debido a su menor tasa de complicaciones a corto y largo plazo186. Están en desarrollo stents biodegradables, fundamentalmente para niños, cuando la aorta todavía tiene que crecer.
Para los adultos, la angioplastia con balón solo está indicada para la redilatación de una aorta con stent previo.
Las técnicas de intervención pediátrica incluyen resección y anastomosis terminoterminal, resección y anastomosis terminoterminal extendida, aortoplastia con parche protésico, aortoplastia con colgajo de subclavia, interposición de un injerto (tubular) y revascularización aortocoronaria con injerto, estas 2 últimas difíciles de practicar en adultoos. La reparación de la recoartación aórtica en adultos puede ser complicada, y los conductos de la aorta ascendente a descendente quizá sean preferibles en casos de anatomía difícil. Aunque el riesgo quirúrgico en la CoA simple hoy sea ≤ 1%, aumenta considerablemente después de los 30-40 años de edad. El riesgo de lesión de la médula espinal es muy bajo187.
Puesto que la CoA no es una enfermedad localizada de la aorta, deben considerarse las complicaciones secundarias que pueden requerir intervención:
- •
Estenosis de la válvula aórtica o insuficiencia secundaria importante.
- •
Aneurisma de la aorta ascendente con un diámetro > 50mm o progresión rápida.
- •
Aneurisma en el punto de CoA anterior.
- •
Aneurismas sintomáticos o grandes en el polígono de Willis.
El tratamiento debe realizarse en centros con amplia experiencia en el tratamiento de CC.
Recomendaciones para la intervención en la coartación y recoartación aórtica
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Está indicada la reparación de la coartación o recoartación aórtica de los pacientes hipertensosc con aumento de la presión no invasiva entre las extremidades superiores e inferiores confirmada por una determinación invasiva (pico-pico ≥ 20 mmHg), preferiblemente percutánea (implante de stent) cuando sea técnicamente factible | I | C |
| Se debe considerar la intervención percutánea (implante de stent) cuando sea técnicamente factible, fuera cual fuere el gradiente de presión, para los pacientes hipertensosc con estrechamiento aórtico ≥ 50% del diámetro aórtico a nivel del diafragma | IIa | C |
| Tengan hipertensión o noc, se debe considerar la intervención percutánea (implante de stent) cuando sea técnicamente factible para los pacientes con aumento de la presión no invasiva confirmada por una determinación invasiva (pico-pico ≥ 20 mmHg) | IIa | C |
| Con independencia del gradiente de presión (gradiente invasivo pico-pico < 20 mmHg) y hipertensiónc, se podría considerar la intervención percutánea (implante de stent) cuando sea técnicamente factible para los pacientes con estrechamiento aórtico ≥ 50% del diámetro aórtico a nivel del diafragma | IIb | C |
aClase de recomendación.
bNivel de evidencia.
cSe debe considerar la monitorización ambulatoria de la presión arterial del brazo derecho para el diagnóstico de hipertensión.
Los residuos, las secuelas y las complicaciones se enumeran a continuación:
- •
La hipertensión arterial en reposo o durante el ejercicio es común, incluso después del éxito del tratamiento, y es un importante factor de riesgo de EC prematura, disfunción ventricular y rotura de aneurismas aórticos o cerebrales188.
- •
La geometría del arco (gótico, almena o normal) y un tamaño aórtico pequeño en la zona del stent podrían participar en el desarrollo de la hipertensión. La determinación ambulatoria de la presión arterial de 24 h en el brazo derecho mejora el diagnóstico de la hipertensión respecto a las determinaciones aisladas189. La relevancia de la hipertensión aislada e inducida por el ejercicio es tema de debate.
- •
Un gradiente de presión sistólica ≥ 20mmHg entre las extremidades superiores y las inferiores indica recoartación y justifica una evaluación invasiva para confirmar el diagnóstico y establecer el tratamiento.
- •
El tratamiento médico de la hipertensión debe seguir las indicaciones de la guía de la ESC/ESH de 2018190.
- •
La CoA recurrente o residual podría inducir o agravar la hipertensión arterial sistémica y sus consecuencias.
- •
Los aneurismas de la aorta ascendente o en el punto de intervención suponen riesgo de rotura y muerte. Las reparaciones con parche, por ejemplo con parche de dacrón, tienen un riesgo especial de aneurisma en el punto de reparación y deben examinarse por imagen con regularidad191.
- •
Es necesario prestar atención a la VAB, la enfermedad de la válvula mitral, la EC prematura y los aneurismas en forma de baya en el polígono de Willis; hoy en día, la mayoría de los médicos no ven indicación para realizar exploraciones sistemáticas a pacientes asintomáticos.
Todos los pacientes con coartación necesitan un seguimiento anual regular. Las imágenes de la aorta, preferiblemente de RMC, son necesarias para documentar la anatomía y las complicaciones (reestenosis o formación de aneurismas) tras la reparación o la intervención. Los intervalos entre las exploraciones dependen de la afección basal y suelen ser cada 3-5 años.
4.6.6Consideraciones adicionales- •
Ejercicio/deporte: los pacientes sin obstrucción residual, normotensos en reposo y durante el ejercicio en general pueden llevar vidas normalmente activas sin restricción. Los pacientes con hipertensión arterial, obstrucción residual u otras complicaciones deben evitar los ejercicios isométricos intensos, en proporción a la gravedad de sus complicaciones.
- •
Embarazo: tras el tratamiento exitoso de la CoA, muchas mujeres toleran el embarazo sin mayores complicaciones43. En concreto, las mujeres con CoA no reparada, pero también aquellas con hipertensión arterial, CoA residual o aneurismas aórticos tras la reparación tienen mayor riesgo de rotura aórtica y rotura de aneurisma cerebral durante el embarazo y el parto. Se ha constatado un exceso de abortos naturales y trastornos hipertensivos192 (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda solo pacientes en alto riesgo (véase el apartado 3.4.6).
El síndrome de Marfan es el prototipo de entidad sindrómica dentro del grupo de EHAT, que comprende un grupo de trastornos de clínica y genética heterogéneas, con aneurisma o disección de la aorta torácica como denominador común. Tanto las formas sindrómicas de EHAT como las no sindrómicas o aisladas forman parte del espectro clínico, con un solapamiento importante entre las distintas entidades.
Para conocer más detalles sobre los distintos síndromes, véase la guía de la ESC de 2014 sobre enfermedades de la aorta193 y el documento de consenso de expertos sobre pruebas genéticas en las CCA/EHAT140. Puesto que la mayoría de los niños con EHAT sobre todo sindrómica van a ser derivados a una unidad de CCA en la edad adulta, la presente guía se centra específicamente en las complicaciones cardiovasculares. El síndrome de Marfan se considera la enfermedad modelo; se mencionan otros síndromes en caso de diferencias importantes con el síndrome de Marfan.
4.7.1.2Presentación clínica e historia naturalAunque la enfermedad de la aorta torácica (ya sean aneurismas detectados mediante cribado o una disección aguda) es la principal característica del síndrome de Marfan/EHAT, la presencia de características extraaórticas en el sistema esquelético/ocular puede ser clave para el diagnóstico.
En esencia, el pronóstico de las EHAT se determina por la dilatación progresiva de la aorta hasta su disección o rotura, las principales causas de muerte. La supervivencia media de los pacientes no tratados es de 40 años, pero la de los pacientes con diagnóstico confirmado puede aproximarse a la de la población general con el tratamiento adecuado29,194. Otras causas cardiovasculares de muerte menos frecuentes son la insuficiencia cardiaca y la MSC29.
En el síndrome de Marfan, el riesgo de disección tipo A aumenta claramente cuando el diámetro de la raíz aórtica es ≥ 50mm195. Otros factores de riesgo son la historia familiar de disección aórtica con diámetros bajos193, la tasa de crecimiento de la raíz aórtica, el embarazo y la hipertensión. Cada vez hay más evidencia de diferencias genéticas en el riesgo de disección. Es posible que también se dilaten otras partes de la aorta.
La presencia de IAo, IT o insuficiencia mitral importantes (normalmente por prolapso valvular) podría causar síntomas de sobrecarga de volumen ventricular, pero la enfermedad del VI puede ocurrir independientemente y acompañarse de arritmia. El prolapso mitral en pacientes con síndrome de Marfan aparece pronto y progresa a insuficiencia grave, necesidad de cirugía y EI más rápidamente que el prolapso mitral idiopático196.
4.7.1.3Estudio diagnósticoEs esencial identificar y establecer tempranamente el diagnóstico, ya que la cirugía profiláctica puede prevenir la disección y la rotura aórticas. Sin duda, el diagnóstico correcto necesita un equipo multidisciplinario capaz de integrar los hallazgos clínicos y genéticos197. Actualmente el diagnóstico del síndrome de Marfan se basa principalmente en los criterios de Ghent, que exigen la aparición de aneurisma/disección de la raíz aórtica y ectopia lentis como características cardinales198. Los criterios para las otras entidades de tipo EHAT están peor definidos.
La identificación del panel de genes es útil para confirmar el diagnóstico y guiar el tratamiento. La tasa de captación de mutaciones en las formas sindrómicas es más alta (> 90%) que en las entidades no sindrómicas (20-30%)199. Una vez identificada una variante patogénica, es obligatorio un cribado genético presintomático de los miembros de la familia que permita establecer el tratamiento precoz.
- •
La evaluación ecocardiográfica de la raíz aórtica debe incluir, además de la determinación del diámetro máximo, las mediciones a nivel de anillo, seno, unión sinotubular y de la aorta ascendente distal, arco y aorta torácica descendente. En los adultos, se recomienda hacer la determinación al final de la diástole con el criterio de «borde principal a borde principal». Los valores obtenidos deben corregirse por edad, sexo y tamaño corporal con nomogramas estándares200,201. Se debe evaluar la morfología (prolapso de la válvula mitral, VAB) y la función valvular, así como la presencia de DAP. La dimensión y la función del VI deben determinarse siguiendo las recomendaciones estándar.
- •
La RMC o la TCC desde la cabeza hasta la pelvis debe realizarse a todos los pacientes, ya que proporciona imágenes de toda la aorta, incluidas las dimensiones aórticas más allá de la raíz. Además de los diámetros aórticos, la información sobre el grado de tortuosidad de las arterias aórtica/vertebrales tiene valor diagnóstico y pronóstico202,203.
- •
Se debe monitorizar con Holter a los pacientes sintomáticos, pues pueden producirse arritmias ventriculares, trastornos de la conducción y MSC.
Aunque los ensayos clínicos no han podido demostrar un beneficio de los bloqueadores beta en la mortalidad o la tasa de disección, estos fármacos constituyen el pilar del tratamiento médico de los pacientes con Marfan/EHAT, pues reducen el estrés de la pared y la tasa de crecimiento de la dilatación204. El riguroso tratamiento médico antihipertensivo, cuyo objetivo es conseguir una presión sistólica < 130mmHg (110mmHg en pacientes con disección aórtica), es importante, aunque no hay datos para establecer umbrales absolutos de presión arterial. Los ARA–II, solos o combinados con los bloqueadores beta, no se han demostrado superiores, pero pueden ser un tratamiento alternativo para pacientes con intolerancia a los bloqueadores beta205,206. El tratamiento médico debe continuarse tras la cirugía.
Los metanálisis en curso de los ensayos con tratamientos médicos pueden ayudar a definir subgrupos, por datos genéticos y clínicos, que se beneficien de un tratamiento específico207. Como no hay ensayos clínicos en otras EHAT diferentes del Marfan, el tratamiento médico en estos casos se adopta a partir de los resultados del Marfan.
4.7.1.5Tratamiento quirúrgicoLas indicaciones para la intervención se resumen en la tabla «Recomendaciones para la cirugía aórtica en las aortopatías».
La cirugía profiláctica de la raíz aórtica es el único tratamiento definitivo para prevenir la disección aórtica en el síndrome de Marfan y las EHAT relacionadas. En pacientes con válvulas anatómicamente normales, cuya insuficiencia es consecuencia del anillo dilatado o la disección, las intervenciones de conservación de la válvula con sustitución de la raíz por prótesis de dacrón y con reimplante de las arterias coronarias en la prótesis (intervención de David) se han convertido en las preferidas, con buenos resultados a largo plazo193,208. La cirugía de sustitución con injerto compuesto, generalmente con válvula mecánica, es una alternativa más duradera, pero requiere anticoagulación de por vida. La decisión sobre qué técnica utilizar debe ser individualizada, teniendo en cuenta las preferencias del paciente y la experiencia quirúrgica209.
El síndrome de Marfan y las EHAT se asocian con riesgo de redisección y aneurisma recurrente en la aorta distal, sobre todo en pacientes con disección previa210,211. Con el aumento de la esperanza de vida, estas complicaciones ahora son más frecuentes. La cirugía aórtica sigue siendo el método de referencia para el tratamiento de la enfermedad de la aorta distal, aunque se podrían considerar procedimientos híbridos con colocación de stents endovasculares en casos seleccionados.
4.7.1.6Recomendaciones sobre el seguimientoEl seguimiento regular de por vida exige la colaboración de especialistas con amplia experiencia en un centro especializado. La ecocardiografía y la RMC/TCC son las pruebas principales.
4.7.1.7Consideraciones adicionales- •
Ejercicio/deporte: se debe aconsejar a los pacientes que eviten el ejercicio a plena potencia y los deportes isométricos, de competición y de contacto. Budts et al.24 proponen calcular el riesgo a partir del tamaño aórtico.
- •
Embarazo: en caso de síndrome de Marfan/EHAT confirmado genéticamente, el riesgo de transmisión tanto para varones como para mujeres es del 50%, por lo que es necesario el oportuno asesoramiento genético. Se recomienda encarecidamente que las mujeres con un diámetro aórtico > 45mm eviten gestar antes de la reparación, por el alto riesgo de disección43. Un diámetro aórtico < 40mm no suele suponer un problema, aunque no existe un diámetro completamente seguro. Con una aorta de 40–45mm, tanto el crecimiento aórtico anterior como los antecedentes familiares son importantes para aconsejar el embarazo con o sin reparación aórtica. Incluso después de la reparación de la aorta ascendente, las pacientes con Marfan siguen teniendo un riesgo de disección de la aorta no reparada (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
Se calcula que un 20–84% de los pacientes con VAB sufrirá dilatación de la aorta ascendente212, lo que indica que la VAB debe considerarse una parte del espectro de las valvulo-aortopatías y que el término «enfermedad aórtica bicúspide» podría ser más adecuado. Aunque la contribución relativa de las anomalías intrínsecas/genéticas de la pared y de las alteraciones hemodinámicas es todavía objeto de debate, es probable que ambos factores estén involucrados.
En ausencia de disfunción valvular relevante, la dilatación aórtica en el contexto de la enfermedad aórtica bicúspide suele ser asintomática. Sin embargo, a medida que aumentan los diámetros, el riesgo de disección aguda aumenta. La incidencia de disección en estos pacientes es 8 veces la de la población general, lo que sigue siendo un riesgo bajo en números absolutos (31/100.000 paciente-años)172,176,213, mucho más bajo que en Marfan/EHAT. Los estudios observacionales indican que el resultado clínico de los pacientes con VAB es más parecido al de la población general con aneurismas y se trata de una aortopatía más benigna que la de Marfan/EHAT176,214.
La CoA se relaciona con mayor riesgo de disección215.
Para conocer más detalles sobre el estudio diagnóstico, véase el apartado 4.7.1.3.
Hasta la fecha, no se dispone de evidencia para el tratamiento médico de la dilatación aórtica en la enfermedad aórtica bicúspide, aunque parece razonable considerar los bloqueadores beta o los ARA–II como tratamiento de primera línea para controlar la hipertensión arterial.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la cirugía aórtica en las aortopatías».
Diversos estudios han establecido una incidencia familiar de BAV de un 5–10% de los familiares de primer grado216. Se recomienda el estudio ecocardiográfico de los familiares de primer grado de los pacientes con BAV; también puede ser adecuado para los deportistas, especialmente varones, cuando haya hipertensión. Las variantes patogénicas raras representan menos del 5% de todos los casos de enfermedad aórtica bicúspide, por lo que las pruebas genéticas habituales no están indicadas, pero pueden considerarse en casos familiares140.
No hay datos sobre el riesgo de disección durante el embarazo de mujeres con aorta dilatada. Según la guía de la ESC de 2018 sobre el tratamiento de la enfermedad cardiovascular en el embarazo43, este está contraindicado cuando el diámetro aórtico sea > 50mm.
Para el tratamiento de la IAo, véase la guía de la ESC/EACTS sobre el tratamiento de la cardiopatía valvular25.
4.7.3Síndrome de TurnerEl síndrome de Turner está causado por una monosomía parcial o completa del cromosoma X y ocurre en 1 de cada 2.500 mujeres nacidas vivas217. Se asocia a estatura baja, retraso de la pubertad, disgenesia ovárica, hipogonadismo hipergonadotrópico, infertilidad, malformaciones cardiacas congénitas, diabetes mellitus, osteoporosis y enfermedades autoinmunitarias. El 50% de las mujeres con síndrome de Turner tienen CC, incluida VAB (con una alta incidencia), CoA, conexión venosa pulmonar anómala, VCS izquierda, anomalías del arco aórtico, dilatación de las arterias braquiocefálicas y dilatación aórtica. Debido a que la prevalencia de anomalías cardiovasculares es muy alta, todas las mujeres con síndrome de Turner deben ser evaluadas por un cardiólogo al menos una vez217. Incluso cuando no haya CC, las personas con síndrome de Turner presentan arteriopatía generalizada; de hecho, el síndrome de Turner por sí solo es un factor independiente de riesgo de dilatación de la aorta torácica. Ser produce disección aórtica (tanto la de tipo A como B) en 40/100.000 personas-años, en comparación con las 6/100.000 personas-años de la población general218.
Para conocer más detalles sobre el estudio diagnóstico, véase el apartado 4.7.1.3.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la cirugía aórtica en las aortopatías».
Con los avances en las técnicas de reproducción asistida y la donación de óvulos, un número creciente de mujeres con síndrome de Turner son capaces de gestar. La presencia de dilatación aórtica y CC aumenta los riesgos del embarazo; además, las mujeres con síndrome de Turner tienen mayor riesgo de sufrir trastornos hipertensivos, incluida la preeclampsia. Todas las mujeres con síndrome de Turner deben recibir asesoramiento sobre el riesgo cardiovascular del embarazo y del tratamiento de fertilidad43.
Recomendaciones para la cirugía aórtica en las aortopatías
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Síndrome de Marfan y EHAT | ||
| Está indicada la reparación de la válvula aórtica, con reimplante o remodelado mediante técnica de anuloplastia aórtica, para los pacientes jóvenes con síndrome da Marfan o EHAT relacionada con dilatación de la raíz aórtica y válvula aórtica tricúspide, siempre que la realicen cirujanos experimentados | I | C |
| Está indicada la cirugía en pacientes con síndrome de Marfan que tengan enfermedad de la raíz aórtica con un diámetro máximo del seno aórtico ≥ 50mmc | I | C |
| Se puede considerar la cirugía para los pacientes con síndrome de Marfan que tengan enfermedad de la raíz aórtica con un diámetro máximo del seno aórtico ≥ 45mmc y otros factores de riesgod | IIa | C |
| Se puede considerar la cirugía para los pacientes con mutaciones en TGFBR1 o TGFBR2 (incluido el síndrome de Loeys Dietz) y enfermedad de la raíz aórtica con un diámetro máximo del seno aórtico ≥ 45mmc | IIa | C |
| Enfermedad de la aorta bicúspide | ||
| Se debe considerar la cirugía aórtica si la aorta ascendente es: | IIa | C |
| • ≥ 50mm en presencia de una válvula bicúspide y otros factores de riesgoe o coartación | ||
| • ≥ 55mm en los demás casos | ||
| Síndrome de Turner | ||
| Se debe considerar la cirugía electiva para los aneurismas de la raíz aórtica o la aorta ascendente de las mujeres con síndrome de Turner mayores de 16 años que tengan un índice de tamaño aórtico > 25 mm/m2 de ASC y factores de riesgo de disecciónf | IIa | C |
| Se puede considerar la cirugía electiva para los aneurismas de la raíz aórtica o la aorta ascendente de las mujeres con síndrome de Turner mayores de 16 años que tengan un índice de tamaño aórtico > 25 mm/m2 de ASC y no presenten factores de riesgo de disecciónf | IIb | C |
ASC: área de superficie corporal; CoA: coartación aórtica; ECG: electrocardiograma; EHAT: enfermedades hereditarias de la aorta torácica; IAo: insuficiencia aórtica; VAB: válvula aórtica bicúspide.
aClase de recomendación.
bNivel de evidencia.
cPara valores de ASC extremos, los puntos de corte recomendados pueden requerir un ajuste.
dHistoria familiar de disección aórtica a diámetro pequeño (o antecedentes personales de disección vascular espontánea), IAo progresiva, intención de gestar, hipertensión no controlada o aumento del tamaño de la aorta > 3mm/año (en mediciones repetidas con la misma técnica de imagen sincronizada con ECG al mismo nivel de la aorta y confirmado por otra técnica).
eHistoria familiar de disección aórtica con diámetro pequeño, intención de gestar, hipertensión sistémica o aumento del tamaño de la aorta > 3mm/año (en mediciones repetidas con la misma técnica de imagen sincronizada con ECG al mismo nivel de la aorta y confirmado por otra técnica).
fVAB, elongación aórtica, CoA o hipertensión.
La OTSVD puede ser subinfundibular, infundibular, valvular o supravalvular.
- •
La estenosis subinfundibular o VD de doble cámara (VDDC) suele estar relacionada con un DSV. Es consecuencia del estrechamiento entre las bandas o líneas musculares prominentes e hipertrofiadas que separan las porciones de entrada y apicales hipertrofiadas de alta presión de las porciones infundibulares de baja presión no hipertrofiadas ni obstructivas del VD219.
- •
La estenosis infundibular suele ocurrir junto con otras lesiones, sobre todo DSV, TdF y secundaria a estenosis pulmonar (EP) valvular (hipertrofia miocárdica reactiva). A nivel infundibular, y en cierta medida a nivel subinfundibular, la obstrucción tiende a ser dinámica, lo que indica que el orificio se estrecha durante la sístole.
- •
La EP valvular suele ser una lesión aislada. Podría haber dilatación del tronco pulmonar y de la AP, principalmente debida a las anomalías intrínsecas de la pared e independiente de la hemodinámica. Lo más común es una válvula pulmonar típica en forma de cúpula con una abertura central estrecha, pero una base de la válvula móvil cardiaca conservada. La válvula pulmonar displásica, con cúspides de escasa movilidad y engrosamiento del tejido mixomatoso, es menos común (entre el 15 y el 20% e incluso menos en adultos no tratados) y con frecuencia forma parte del síndrome de Noonan. En los adultos, la válvula pulmonar estenótica puede calcificarse con el paso del tiempo.
- •
La EP supravalvular o estenosis arterial pulmonar es consecuencia del estrechamiento del tronco pulmonar principal, la bifurcación arterial pulmonar o las ramas pulmonares. Casi nunca ocurre sola, y puede darse en la TdF, el síndrome de Williams-Beuren, el síndrome de Noonan, el síndrome de Keutel, el síndrome de la rubéola congénita o el síndrome de Alagille. La estenosis puede localizarse en las ramas principales o ser más periférica; puede ser discreta o difusa (hipoplásica) o con oclusión franca, y ser única o múltiple. La estenosis puede ser secundaria a la colocación previa de una banda en la AP o en un punto de cortocircuito anterior. Un diámetro de estenosis ≥ 50% suele considerarse relevante, y se espera que tenga un gradiente de presión y resulte en hipertensión de la AP proximal.
- •
Subinfundibular/infundibular: los pacientes adultos con VDDC no intervenido pueden estar asintomáticos o presentar angina, disnea, mareos o síncope. El grado de obstrucción avanza con el tiempo220.
- •
Valvular: los pacientes con EP valvular de leve a moderada suelen estar asintomáticos. La EP valvular leve en adultos no intervenidos suele ser no progresiva221. La EP moderada valvular (calcificación) o subvalvular puede avanzar por la hipertrofia miocárdica reactiva. Los pacientes con estenosis grave pueden tener disnea, capacidad de ejercicio reducida y peor pronóstico.
- •
Supravalvular: los pacientes pueden estar asintomáticos o tener síntomas de disnea y capacidad de ejercicio reducida. Se suele reconocerlos en el contexto de ciertos síndromes o al derivarlos por sospecha de hipertensión pulmonar. La estenosis de la AP periférica puede avanzar en gravedad.
Véase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos incluyen soplo sistólico fuerte en la obstrucción y desdoblamiento del segundo ruido cardiaco. En la EP periférica, el soplo sistólico suele oírse por encima de los campos pulmonares.
- •
La ecocardiografía es la técnica diagnóstica de primera línea, ya que proporciona la visualización del nivel de la OTSVD, la anatomía de la válvula pulmonar, la AP y las ramas proximales. La ecocardiografía Doppler permite cuantificar las velocidades de flujo a lo largo de la obstrucción y evaluar la gravedad. La correlación entre las velocidades de flujo y los gradientes de presión solo es fiable en caso de estenosis discreta, por ejemplo, EP valvular aislada. En presencia de función normal del VD y flujo transvalvular normal, la OTSVD se considera leve cuando el gradiente máximo a lo largo de la obstrucción es < 36mmHg, moderada entre 36 y 64mmHg y grave cuando el gradiente es > 64mmHg. En caso de estrechamiento alargado o cuando haya varias estenosis en serie (subvalvular y valvular), la ecuación de Bernoully puede sobrestimar el gradiente de presión. La velocidad del flujo por Doppler en la IAo proporciona un cálculo más fiable de la presión del VD y la gravedad de la OTSVD. Los gradientes solo representan la gravedad de una obstrucción si hay buena función sistólica del VD. En una situación de bajo flujo y bajo gradiente, es muy difícil evaluar la gravedad de la OTSVD222.
- •
La RMC y la TCC suelen proporcionar información adicional importante al identificar los lugares de obstrucción, sobre todo a nivel subinfundibular, del conducto o de las ramas de la AP, así como la evaluación del VD, el anillo pulmonar, las dimensiones del tracto de salida y de la arteria y el flujo pulmonar diferencial. Son los métodos de elección para visualizar la dilatación pulmonar y la EP periférica.
- •
El cateterismo cardiaco podría requerirse para confirmar el alcance, la gravedad y el nivel de la obstrucción, como por ejemplo el VDDC.
Se recomienda la valvulotomía percutánea con balón para los pacientes con EP valvular con válvulas no displásicas o con EP periférica (a menudo con implante de stent)223. La cirugía está recomendada para los pacientes con EP subinfundibular o infundibular y anillo pulmonar hipoplásico, con válvulas pulmonares, y pacientes con lesiones asociadas que necesitan tratamiento quirúrgico, como insuficiencia pulmonar (IP) grave o IT grave. La EP rara vez se puede tratar con cirugía.
Tanto las cirugías como los cateterismos deben realizarse únicamente en centros especializados en CC.
Los pacientes con EP subvalvular, valvular y supravalvular pueden tener un tronco pulmonar visiblemente dilatado. La rotura es muy poco común en estos vasos de baja presión y muy elásticos, por lo que en general estos aneurismas no requieren intervención224.
Para el conducto VD-AP, véase el apartado 4.14.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención en la obstrucción del tracto de salida del ventrículo derecho» y la figura 7.
Recomendaciones para la intervención en la obstrucción del tracto de salida del ventrículo derecho
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| La valvuloplastia con balón es la intervención de elección para la EP si es anatómicamente factible | I | C |
| La OTSVD, a cualquier nivel, debe repararse con independencia de los síntomas cuando el gradiente Doppler máximo sea > 64mmHgc siempre que la función del VD sea normal y no se necesite sustitución valvular | I | C |
| Cuando la sustitución quirúrgica de la válvula sea la única opción para el paciente con estenosis grave sintomático, la cirugía está indicadad | I | C |
| Cuando la sustitución quirúrgica de la válvula sea la única opciónd para el paciente con estenosis grave asintomático, la cirugía está indicada en presencia de:• Reducción objetiva de la capacidad de ejercicio o• Función del VD decreciente o progresión de la IT a moderada o más• Presión sistólica del VD > 80 mmHg• Cortocircuito D-I por CIA o DSV | I | C |
| Se debe considerar intervenir a los pacientes con un gradiente Doppler máximo < 64mmHg en presencia de:• Síntomas relacionados con la EP o• Función del VD decreciente o progresión de la IT a moderada o más• Cortocircuito D-I por CIA o DSV | IIa | C |
| Se debe considerar la reparación de la EP periférica, con independencia de los síntomas, si hay un estrechamiento del diámetro > 50% y presión sistólica del VD > 50mmHg o anomalías en la perfusión pulmonar | IIa | C |
CIA: comunicación interauricular; DSV: defecto del septo ventricular; EP: estenosis pulmonar; D-I: derecha-izquierda; IT: insuficiencia tricuspídea; OTSVD: obstrucción del tracto de salida del ventrículo derecho; VD: ventrículo derecho.
aClase de recomendación.
bNivel de evidencia.
cLa presión sistólica del VD estimada a partir de la velocidad de la IT debe confirmar la EP grave.
dEl umbral para la intervención es más alto cuando se requiere sustitución valvular, porque se debe tener en cuenta los riesgos a largo plazo, como la endocarditis y la necesidad de reintervención por fallo de la válvula protésica.
 CIA: comunicación interauricular;
CIA: comunicación interauricular; Tratamiento de la obstrucción del tracto de salida del ventrículo derecho. CIA: comunicación interauricular; D-I: derecha-izquierda; DSV: defecto del septo ventricular; EP: estenosis pulmonar; IT: insuficiencia tricuspídea; OTSVD: obstrucción del tracto de salida del ventrículo derecho; PSVD: presión sistólica ventricular derecha; VD: ventrículo derecho.
aPara la EP periférica, con independencia de los síntomas, debería considerarse la reparación si hay un estrechamiento del diámetro > 50% y una PSVD > 50mmHg y/o anomalías en la perfusión pulmonar.
bLa valvuloplastia con balón es la intervención de elección para la EP si es anatómicamente factible.
Los pacientes con OTSVD necesitan un seguimiento ecocardiográfico regular de por vida. La frecuencia del seguimiento depende de la gravedad de la lesión, pero la mayoría de los pacientes necesitarán una visita anual que incluya evaluación en centros especializados en CCA. Después de la reparación, la persistencia de la IP puede requerir una reintervención posterior si el paciente se vuelve sintomático o aparece dilatación o disfunción progresiva del VD (véase el apartado 4.10). Los pacientes con EP valvular leve o residual leve solo necesitan un seguimiento cada 5 años.
4.8.6Consideraciones adicionales- •
Ejercicio/deporte: no hay restricciones para pacientes con EP (residual) leve. Los pacientes con EP moderada deben evitar los deportes estáticos y de competición. Los pacientes con EP grave deben limitarse a los deportes de baja intensidad.
- •
Embarazo: se tolera bien a menos que la OTSVD sea demasiado grave o que la insuficiencia del VD sea una cuestión considerable. La valvulotomía percutánea con balón puede realizarse durante el embarazo, pero pocas veces es necesaria (véase el apartado 3.5.7).
- •
Profilaxis contra EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
La anomalía de Ebstein, una enfermedad relativamente rara, se caracteriza por la formación anómala y el desplazamiento apical de las valvas de la válvula tricúspide. La valva anterior suele originarse de forma adecuada a nivel anular, pero está dilatada y con forma de vela, mientras las valvas septal y posterior están desplazadas hacia el vértice ventricular derecho y a menudo ligadas al endocardio.
El desplazamiento apical de la válvula tricúspide implica que el corazón derecho consta de una AD, una porción auricularizada del VD y el restante VD, funcional. La válvula tricúspide suele ser regurgitante.
Las anomalías asociadas más frecuentes incluyen cortocircuito a nivel auricular (DSA del ostium secundum o foramen oval permeable) y vías accesorias múltiples, incluidas las de tipo Mahaim. La presencia de vías accesorias múltiples junto con TA y FA se asocia con MSC. Se halla anomalía de la válvula tricúspide sistémica compatible con malformación de Ebstein en 1 tercio de las TGAcc.
Los cambios hemodinámicos dependen de la gravedad de la disfunción de la válvula tricúspide, el grado de auricularización del VD, la contractilidad del restante ventrículo funcional y el ventrículo sistémico, el tipo y la gravedad de las anomalías concomitantes y las arritmias.
La fisiopatología se caracteriza por regurgitación sistólica de sangre desde el VD funcional a través de la válvula tricúspide hacia el ventrículo auricularizado o la AD, que tiende a dilatarse. Una CIA permite un cortocircuito I-D o, sobre todo durante el ejercicio, D-I. La anomalía de Ebstein podría resultar en un rendimiento cardiaco sistémico crónicamente bajo.
4.9.2Presentación clínica e historia naturalLa presentación clínica varía desde síntomas triviales hasta un defecto cardiaco cianótico profundo. Los pacientes con formas leves pueden estar asintomáticos durante décadas hasta el diagnóstico. Las complicaciones típicas incluyen IT de alto grado, disfunción del VD, insuficiencia del VD, cirrosis hepática, abscesos cerebrales, embolia paradójica, embolia pulmonar, taquiarritmias, MSC y EI. Los síntomas clave son arritmias (la taquicardia por reentrada auriculoventricular [TRAV] es la más frecuente), disnea, fatiga, mala tolerancia al ejercicio, dolor pectoral y cianosis periférica o central.
4.9.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos podrían incluir cianosis y hepatomegalia. Los hallazgos auscultatorios incluyen desdoblamiento del primer y el segundo ruido cardiaco, soplos en serie, tercer y cuarto ruido cardiaco, triple o cuádruple ritmo y un soplo sistólico de la IT. El ECG podría revelar hipertrofia auricular derecha, un intervalo PR prolongado, bloqueo de rama derecha, a menudo con complejo QRS fragmentado, Q profunda en II, III, F y V1-V4, síndrome de preexcitación, presión baja y arritmias supraventriculares y ventriculares.
- •
La radiografía es útil para seguir los cambios en el tamaño del corazón.
- •
La ecocardiografía es la técnica diagnóstica clave, ya que ofrece información sobre la anatomía y la función de la válvula tricúspide, el desplazamiento distal apical de la valva septal o posterior (en adultos, área de superficie corporal ≥ 0,8cm/m2), tamaño de la valva anterior, anclaje de la valva de la válvula tricúspide septal o posterior sobre el septo o la pared ventricular, el tamaño y la función de las diferentes secciones cardiacas (AR, ventrículo auricularizado, VD funcional restante y VI), OTSVD y lesiones asociadas.
- •
La RMC es valiosa para establecer el pronóstico225 y la evaluación con vistas a la cirugía, ya que ofrece perspectivas ilimitadas para examinar el corazón derecho dilatado y la función del VD y la válvula tricúspide.
Los síntomas clínicos determinan el tratamiento. Un tratamiento conservador puede aliviar los síntomas temporalmente y crear una base beneficiosa para la posterior intervención226. La anticoagulación oral se recomienda para pacientes con antecedentes de embolia paradójica o FA. Se puede considerar la anticoagulación oral en caso de mayor riesgo de tromboembolia o cortocircuito D-I. Los trastornos sintomáticos del ritmo se pueden tratar de forma conservadora o, preferiblemente, mediante ablación227. El acceso transcatéter a las vías accesorias del lado derecho y la vía lenta del nódulo AV puede verse obstaculizado por la cirugía de la válvula tricúspide, por lo que puede ser razonable evaluar los sustratos arritmogénicos y, si se identifican, proceder a la ablación antes de la cirugía. A veces puede haber indicación para cerrar solo la comunicación auricular. No obstante, esto debe discutirse cuidadosamente, ya que podría producir un mayor aumento en las presiones del corazón derecho y un descenso en el gasto cardiaco sistémico. La reparación quirúrgica sigue siendo un desafío y solo deben realizarla cirujanos con experiencia concreta en esta lesión. Si es factible, se prefiere la reparación de la válvula tricúspide antes que la sustitución (con cierre de una CIA asociada). La anastomosis cavopulmonar bidireccional adicional puede ser necesaria si el VD es demasiado pequeño para la corrección o si se ha desarrollado disfunción del VD228. Para los pacientes con reparación fallida o en disfunción biventricular grave, el trasplante cardiaco podría ser la única opción.
La mortalidad operatoria, que antes era alta (> 25%), ha caído por debajo del 6% en centros especializados. Más del 90% de los pacientes intervenidos por un cirujano con experiencia sobreviven más de 10 años, muchos de ellos en clase funcional I-II. Las mortalidades tardías probablemente se deban a las arritmias. En una extensa serie, las supervivencias sin reintervención tardía fueron del 86, el 74, el 62 y el 46% a los 5, 10, 15 y 20 años respectivamente229.Extract from original table on pag. 49 From ‘Original version.pdf’ file
Recomendaciones para la intervención en la anomalía de Ebstein
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Indicaciones para la cirugía | ||
| La reparación quirúrgica está indicada para los pacientes sintomáticos con IT grave o capacidad de ejercicio deteriorada medida por PECP | I | C |
| La reparación quirúrgica debe realizarla un cirujano especialista en CC con experiencia específica en cirugía de Ebstein | I | C |
| Si también hay una indicación para cirugía de la VT, el cierre de CIA/FOP debe ser quirúrgica al tiempo de reparar la válvula si la hemodinámica lo tolera | I | C |
| Con independencia de los síntomas, se debe considerar la reparación quirúrgica para los pacientes con dilatación progresiva del corazón derecho o reducción de la función sistólica del VD | IIa | C |
| Indicaciones para el cateterismo intervencionista | ||
| Los pacientes con arritmias relevantes deben ser sometidos a prueba electrofisiológica, seguida de terapia con ablación, de ser posible, o tratamiento quirúrgico de las arritmias en caso de cirugía cardiaca programada | I | C |
| En caso de embolia sistémica documentada probablemente causada por embolia paradójica, debe considerarse el cierre solo con dispositivo de CIA/FOP, pero requiere evaluación cuidadosa antes de la intervención para excluir inducción del aumento de presión de la AD o caída del gasto cardiaco | IIa | C |
| Si la cianosis (saturación de oxígeno < 90% en reposo) es la complicación principal, podría considerarse el cierre aislado con dispositivo de DSA/FOP, pero requiere evaluación cuidadosa antes de la intervención para excluir inducción del aumento de presión de la AD o caída del gasto cardiaco | IIb | C |
AD: aurícula derecha; CC: cardiopatías congénitas; CIA: comunicación interauricular; ECG: electrocardiograma; FOP: foramen oval permeable; PECP: prueba de esfuerzo cardiopulmonar; IT: insuficiencia tricuspídea; VD: ventrículo derecho; VT: válvula tricúspide.
aClase de recomendación.
bNivel de evidencia.
Todos los pacientes necesitan seguimiento regular al menos 1 vez al año en centros especializados en CCA. Las anomalías residuales posoperatorias típicas que buscar son IT persistente o nueva, las complicaciones habituales tras sustitución de la válvula, insuficiencia del VD o el VI, cortocircuito auricular residual, arritmias y bloqueos cardiacos de grado más alto.
Podría ser necesario reintervenir en caso de IT recurrente e insuficiencia de válvulas protésicas.
4.9.6Consideraciones adicionales- •
Ejercicio/deporte: los pacientes sin anomalías residuales en general pueden llevar vidas normalmente activas sin restricción, con la excepción de los deportes estáticos de competición exhaustivos. Los pacientes con IT más que leve, disfunción ventricular, cortocircuito, arritmias u otras complicaciones deben evitar los ejercicios isométricos intensos, en proporción a la gravedad de sus complicaciones.
- •
Embarazo: las mujeres asintomáticas con buena función ventricular pueden tolerar bien el embarazo. Hay cierto riesgo de insuficiencia del VD, arritmia y embolia paradójica. El embarazo será de riesgo más alto en presencia de cianosis relevante, arritmia grave e insuficiencia cardiaca derecha (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
La TdF es consecuencia de la desviación anterocefálica del septo de salida, que resulta en las 4 características siguientes: DSV no restrictivo, cabalgamiento aórtico (pero < 50%), OTSVD que puede ser infundibular, valvular o, por lo general, una combinación de ambos, con o sin estenosis supravalvular o de las ramas de la AP, y consiguiente HVD. Las poblaciones con TdF se subdividen en pacientes sindrómicos (aproximadamente el 20%, tales como: microdeleción del cromosoma 22q11, trisomía 21 y síndromes de Alagille, Noonan, Williams y Klippel Feil) y pacientes no sindrómicos (la gran mayoría)230. La tasa de mortalidad estándar entre los pacientes con TdF reparada es casi el doble que la de los pacientes con defectos simples (CIA y DSV)231.
4.10.2Presentación clínica e historia naturalLa reparación quirúrgica de la TdF ha cambiado a lo largo del tiempo y supone el cierre del DSV y alivio de la OTSVD, con resección del infundíbulo y valvulotomía pulmonar; muchos pacientes necesitan parches transanulares para agrandar el paso entre el VD y la AP. En algunos casos se hace un procedimiento paliativo del cortocircuito para aumentar el flujo sanguíneo pulmonar antes de la reparación. Las complicaciones comunes en la edad adulta son:
- •
IP: casi siempre hay IP importante tras una reparación con parche transanular. Normalmente, la IP se tolera bien durante años. No obstante, la IP crónica grave con el tiempo produce dilatación y disfunción sintomáticas del VD232. La gravedad de la IP y sus efectos deletéreos a largo plazo aumentan con la estenosis distal de la AP o la HAP.
- •
OTSVD residual: puede ocurrir en el infundíbulo, a nivel de la válvula pulmonar y el tronco pulmonar principal, distalmente, más allá de la bifurcación, y a veces en las ramas de las AP izquierda y derecha. El aumento de la presión del VD y la HVD son factores independientes de riesgo de mal pronóstico y poca capacidad de ejercicio a pesar de un menor volumen del VD233.
- •
DSV residual: puede deberse a la dehiscencia parcial del parche o un cierre completo no logrado en la cirugía; podría producir sobrecarga de volumen del VI.
- •
Complicaciones aórticas: pueden aparecer muchos años después de la reparación inicial e incluyen la dilatación de la raíz aórtica progresiva e IAo (rara vez disección aórtica). El mecanismo subyacente no se conoce bien y puede estar relacionado con la dilatación de la aorta ascendente, una elasticidad aórtica anómala y el tipo de reparación quirúrgica234.
- •
Disfunción del VI y el VD/insuficiencia cardiaca: la dilatación del VD suele ser consecuencia de una IP de larga duración en presencia o ausencia de OTSVD. Puede haber IT significativa como consecuencia de la dilatación del VD, lo que a su vez empeora la dilatación del VD. La dilatación del VI puede aparecer como consecuencia de cortocircuitos arteriales paliativos de larga duración, DSV residuales o IAo. La disfunción del VD y del VI puede aparecer por la cianosis de larga duración antes de la reparación o la protección miocárdica inadecuada durante la reparación. También puede resultar de una interacción ventriculoventricular adversa, desincronía electromecánica235,236 y anomalías coronarias. Se ha descrito una deformación longitudinal de la pared libre del VI reducida a pesar de la FEVI conservada237. La incidencia de insuficiencia cardiaca clínica con signos y síntomas típicos aumenta significativamente con la edad238. Los mecanismos subyacentes pueden ser: daño miocárdico, secuelas de la reparación quirúrgica o trastornos de la conducción eléctrica. Las estrategias de tratamiento que son efectivas en la cardiopatía adquirida también suelen aplicarse a estos pacientes, aunque su eficacia en la insuficiencia del VD es incierta239.
- •
Arritmias auriculares/ventriculares y MSC: las arritmias y la MSC son complicaciones tardías importantes. La prevalencia de por vida de las arritmias auriculares es de 20%. La TRIA que involucra el istmo cavotricuspídeo y la incisión de la AD están relacionadas con el agrandamiento de la AD, mientras que la FA se ve facilitada por la dilatación de la AI. Las arritmias ventriculares abarcan la TV/FV polimórfica, típicamente relacionada con función del VD y el VI deterioradas graves, y la TV sostenida monomórfica, que son particularmente relevantes en la TdF34. A pesar de que tanto la TV/FV polimórfica como la TV monomórfica pueden causar MSC, con una frecuencia del 1–3,5% en los estudios retrospectivos32, es necesario conocer los diferentes sustratos arritmogénicos ventriculares subyacentes para la estratificación del riesgo y el tratamiento. La disfunción del VI sistólica o diastólica y las taquiarritmias ventriculares y auriculares son predictivas de muerte y TV sostenida en adultos con TdF240. Los posibles factores de riesgo relacionados con cualquier arritmia ventricular y MSC en la TdF son: duración del QRS ≥ 180ms, disfunción del VI sistólica o diastólica y TV inducible en la prueba EF. La edad avanzada en el momento de la SVP y una HVD con disfunción previa a la intervención pueden ser predictivas de una supervivencia posoperatoria corta y arritmia ventricular sostenida241. Los sustratos dominantes para la TV monomórfica son los istmos anatómicamente definidos rodeados por tejido no excitable. La dimensión del istmo y sus propiedades de conducción pueden evaluarse por mapeo con catéter y suelen determinar la susceptibilidad a las arritmias. La ablación de los istmos anatómicos ha sido muy eficaz para controlar la TV242. Es necesario investigar la utilidad del mapeo con catéter en la estratificación individualizada del riesgo.
- •
–La endocarditis puede aparecer tras la SVP quirúrgica o percutánea. Las prótesis que contienen válvulas son un importante factor independiente de riesgo de EI a corto y largo plazo después del implante, mientras que las prótesis que no contienen válvulas son un factor de riesgo solo durante los primeros 6 meses después del implante76.
En la figura 8 se muestra una descripción esquemática de las complicaciones a largo plazo tras la reparación de la TdF.
 DSV: defecto del septo ventricular; EP: estenosis pulmonar;
DSV: defecto del septo ventricular; EP: estenosis pulmonar; Tratamiento de la tetralogía de Fallot reparada: complicaciones a largo plazo que atender en el seguimiento. AD: aurícula derecha; AI: aurícula izquierda; Ao: aorta; AP: arteria pulmonar; DSV: defecto del septo ventricular; EP: estenosis pulmonar; MSC: muerte súbita cardiaca; VD: ventrículo derecho; VI: ventrículo izquierdo; VT: válvula tricúspide.
Véase el apartado 3.3 para conocer los principios generales.
- •
Los hallazgos clínicos incluyen un desdoblamiento del segundo ruido cardiaco. El soplo diastólico precoz al final de tono grave indica IP grave. El soplo sistólico de eyección de tono agudo y prolongado indica OTSVD; el soplo diastólico de tono agudo inadica IAo y el soplo pansistólico, DSV residual.
- •
El ECG revela, en general, el bloqueo de rama derecha completo; el QRS ancho refleja el grado de dilatación del VD.
- •
La ecocardiografía es la técnica diagnóstica de primera línea, ya que ofrece la evaluación de la OTSVD y la IP residuales, el DSV residual, el tamaño y la función del VD y el VI241, la IT, la presión ventricular derecha (PVD), el tamaño de la raíz aórtica y la IAo. Las mediciones de deformación son útiles para cuantificar el grado de desicronía electromecánica243.
- •
La RMC es el método de elección para la evaluación del volumen y la función del VD, la IP, el tamaño, la forma y la expansión de las AP y la aorta ascendente, el infundíbulo, la posición de las grandes arterias o conductos respecto al esternón (reesternotomía) y el grado de cortocircuito residual. El realce tardío con gadolinio demuestra fibrosis, cuyo alcance se relaciona con otros factores de riesgo de TV y MSC244. El mapeo en T1 puede tener un papel emergente.
- •
La TCC proporciona información sobre arterias coronarias (especialmente importante para la evaluación de la relación espacial con el TSVD previo a la IPVP o la cirugía), el alcance de la calcificación del conducto (anclaje percutáneo de la válvula) y la presencia de colaterales aortopulmonares principales (MAPCA). También puede considerarse como una alternativa a la cuantificación del VD en pacientes que no pueden someterse a RMC.
- •
La PECP ayuda a elegir el momento de la reintervención y ofrece información pronóstica23.
- •
La monitorización Holter, el grabador de eventos y las pruebas EF son necesarias en pacientes seleccionados (en alto riesgo, en estudio por arritmia sospechada o clínica o evaluados para reintervención del TSVD). La TV sostenida inducible tiene valor pronóstico de TV clínica y MSC245.
- •
El cateterismo cardiaco debe limitarse a pacientes sometidos a intervenciones percutáneas, esto es, alivio de la estenosis distal de la AP o implante percutáneo de válvula, y en caso de que la evaluación no invasiva no sea concluyente. Antes de la cirugía, la angiografía coronaria puede visualizar las arterias coronarias, importante para evaluar la relación espacial con el TSVD antes del IPVP.
La sustitución de la válvula pulmonar (SVP) o el alivio de la OTSVD pueden realizarse con una mortalidad baja en pacientes sin insuficiencia cardiaca o disfunción ventricular en fase avanzada246. La IP es el motivo más frecuente para considerar la cirugía. Establecer el momento óptimo sigue siendo un desafío. Los datos longitudinales son más importantes que las mediciones individuales para ayudar a decidir el momento de la reintervención247. La normalización del tamaño del VD tras la reintervención es poco probable si el índice telesistólico excede los 80ml/m2 y el volumen diastólico final supera los 160ml/m2, aunque este valor de corte para la reintervención puede no tener correlación con el beneficio clínico248–250. Un metanálisis reciente ha demostrado que la SVP puede mejorar los síntomas y reducir el volumen del VD, pero todavía no se ha demostrado que mejore la supervivencia251.
La estenosis distal de la AP debe tratarse en el momento de la cirugía (incluido el implante de stent intraoperatorio) o vía percutánea. La SVP con válvula pulmonar biológica (xenoinjerto u homoinjerto) parece tener una vida útil de 10-20 años248,252,253 y en el futuro se podrá realizar la sustitución mediante procedimientos de tipo «válvula-en-válvula». La experiencia con válvulas mecánicas es escasa en este contexto y preocupa una anticoagulación adecuada.
La anuloplastia de la VT, los DSV residuales o la dilatación de la raíz aórtica/IAo deben tratarse también en el momento de la cirugía. La indicación de cirugía de la raíz aórtica es la misma que para la población general254.
Las técnicas de IPVP se han convertido en una alternativa a la cirugía, sobre todo para pacientes con regurgitación o estenosis en el TSVD, aunque también en pacientes seleccionados con estenosis/regurgitación en el TSVD nativo. El IPVP, cuando es técnicamente factible, proporciona resultados comparables a los de la SVP quirúrgica, y tiene como objetivo aumentar la vida útil del conducto y reducir así el número de reoperaciones a las que debe someterse el paciente a lo largo de su vida255. La rotura de un stent (inicialmente, la complicación más habitual) se ha convertido en un problema menor cuando se prepara cuidadosamente la zona con stents adicionales. Los mejores resultados a largo plazo se producen cuando se puede alcanzar un gradiente residual < 15mmHg256. Otras complicaciones poco frecuentes (< 2% de los pacientes) son la rotura del conducto y la compresión de la arteria coronaria. El riesgo de endocarditis tras el IPVP sigue siendo un problema, con una incidencia anual de un 2–3%257,258. Debido a que la compresión de la arteria coronaria puede poner en peligro la vida, se debe realizar una prueba con balón para excluir una posible compresión antes del IPVP, aunque esta prueba conlleva un riesgo de rotura del conducto. En caso de calcificación extensa del conducto circunscrito, el IPVP solo debe realizarse si la exploración por TCC muestra una distancia suficiente entre el conducto y las arterias coronarias. La asociación estrecha que existe entre los istmos anatómicos de conducción lenta y la TV monomórfica sostenida, y la posible pérdida de acceso al istmo anatómico por ablación con catéter después de la SVP quirúrgica o el implante percutáneo de válvulas en los TSVD con parche, tiene importantes implicaciones para los pacientes que se someten a reintervenciones259. Se investiga el posible beneficio del mapeo previo a la intervención y la ablación preventiva de los istmos anatómicos de conducción lenta antes o durante la intervención en pacientes sin TV sostenida espontánea documentada.
Recomendaciones para la reintervención tras reparación de la tetralogía de Fallot
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| La SVP está indicada para pacientes sintomáticos con IP gravec u OTSVD al menos moderadad | I | C |
| Para los pacientes que no tienen tracto de salida nativoe, se prefiere el cateterismo intervencionista (IPVP) si es anatómicamente factible | I | C |
| Se podría considerar la SVP para los pacientes asintomáticos con IP grave u OTSVD cuando tengan al menos 1 de los siguientes criterios:• Reducción objetiva de la capacidad de ejercicio• Dilatación progresiva del VD a VTSVDi ≥ 80 ml/m2 o VTDVDi ≥ 160 ml/m2 o progresión de la IT a ≥ moderadaf• Disfunción sistólica del VD progresiva• OTSVD con presión sistólica del VD > 80 mmHg | IIa | C |
| Se debe considerar el cierre del defecto de los pacientes con DSV y sobrecarga de volumen VI importante o que vayan a someterse a cirugía de la válvula pulmonar | IIa | C |
| Para los pacientes con TV sostenida que vayan a someterse a SVP o implante percutáneo de válvula, debe considerarse el mapeo prequirúrgico con catéter y la resección de los istmos anatómicos relacionados con la TV antes o durante la intervención | IIa | C |
| Se debe considerar una evaluación electrofisiológica que incluya estimulación eléctrica programada para la estratificación del riesgo de MSC de los pacientes con factores de riesgo adicionales (disfunción del VI/VD; TV no sostenida sintomática; QRS ≥ 180ms, cicatrización extensa del VD en la RMC) | IIa | C |
| Se debe considerar la implantación de DAI Para pacientes seleccionados con TdF y múltiples factores de riesgo de MSC, como disfunción del VI, TV no sostenida sintomática, QRS ≥ 180ms, cicatrización del VD extensa en la RMC o TV inducible en la estimulación eléctrica programada | IIa | C |
| Para los pacientes con función biventricular conservada, se puede considerar la ablación con catéter o la ablación quirúrgica concomitante como alternativa al DAI para la TV monomórfica sostenida sintomática siempre que el procedimiento se realice en centros con mucha experiencia y se hayan alcanzado los objetivos previamente establecidos (p. ej., no inducibilidad, bloqueo de la conducción a través de las líneas de ablación) | IIb | C |
DAI: desfibrilador automático implantable; DSV: defecto del septo ventricular; IP: insuficiencia pulmonar; IPVP: implante percutáneo de la válvula pulmonar; IT: insuficiencia tricuspídea; MSC: muerte súbita cardiaca; OTSVD: obstrucción del tracto de salida del ventrículo derecho; RMC: resonancia magnética cardiovascular; SVP: sustitución de válvula pulmonar; TdF: tetralogía de Fallot; TSVD: tracto de salida del ventrículo derecho; TV: taquicardia ventricular; VD: ventrículo derecho; VI: ventrículo izquierdo; VTDVDi: volumen telediastólico del ventrículo derecho indexado; VTSVDi: volumen telesistólico del ventrículo derecho indexado.
aClase de recomendación.
bNivel de evidencia.
cFracción regurgitante por RMC > 30-40%.
dVelocidad pico > 3 m/s.
ePacientes con cirugía previa del TSVD con homoinjertos, injertos bovinos de la vena yugular o bioprótesis/conductos.
fConfirmado mediate determinaciones repetidas.
El DAI debe implantarse para la prevención secundaria de la MSC (pacientes con parada cardiaca o TV sostenida) (recomendación I C). El implante de DAI para la prevención primaria sigue siendo polémico, y de momento no se ha desarrollado ningún esquema ideal de estratificación del riesgo. Los pacientes con síncope inexplicable y función ventricular deteriorada u otros factores de riesgo de MSC deberían someterse a evaluación hemodinámica y EF. En ausencia de una causa definida y reversible, se debe considerar el implante de DAI (véase el apartado 3.4.2)260,261.
4.10.6Recomendaciones sobre el seguimientoTodos los pacientes con TdF deben tener un seguimiento cardiaco periódico en un centro especializado en CCA, que en la mayoría de los casos debería ser anual. La evaluación de seguimiento debe buscar las complicaciones ya enumeradas (véase apartado 4.10.2). Todos los pacientes deberían someterse a RMC. Los intervalos para repetir los estudios dependen de los hallazgos patológicos.
4.10.7Consideraciones adicionales- •
Ejercicio/deporte: no hay restricciones para pacientes asintomáticos con buena hemodinámica. Los pacientes con riesgo alto de arritmia clínica/MSC, disfunción biventricular en fase avanzada o con acusada aortopatía ascendente deberían limitarse a realizar actividades/deportes de baja intensidad y evitar el ejercicio isométrico.
- •
Embarazo: para las pacientes no reparadas, supone un riesgo considerable de complicaciones y muerte tanto maternas como fetales. El riesgo con el embarazo de las pacientes no reparadas depende del estado hemodinámico; es bajo en pacientes con buena hemodinámica. En pacientes con lesiones residuales relevantes, hay riesgo de arritmia e insuficiencia cardiaca derecha, y el embarazo puede tener un efecto secundario a largo plazo en la función cardiovascular262 (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda solo para pacientes en alto riesgo (véase el apartado 3.4.6).
Los pacientes adultos con atresia pulmonar y DSV son una población heterogénea en términos de anatomía, fisiología e intervenciones previas. La atresia pulmonar con DSV comparte la anatomía intracardiaca de la TdF, pero carece de comunicación directa entre el VD y las AP. La microdeleción del 22q11.2 es frecuente (anomalías faciales, voz nasalizada y retraso del desarrollo)263. El grado de complicación con las AP es variable en la atresia pulmonar con DSV y determina tanto la presentación clínica como el tratamiento (la complejidad del lecho vascular pulmonar podría hacer que la reparación resulte menos atractiva e incluso imposible).
El caso de los pacientes con conexiones cardiacas discordantes o una fisiología ventricular única y su tratamiento se discute en los apartados pertinentes.
Hay 3 patrones de AP:
- •
Unifocal con AP confluentes de buen tamaño facilitado por DAP.
- •
Multifocal, con AP confluentes pero hipoplásicas (aspecto de gaviota) facilitado por múltiples MAPCA.
- •
Multifocal con AP no confluentes facilitado por MAPCA.
El tratamiento quirúrgico es un tema muy controvertido debido a la falta de consenso sobre el tratamiento óptimo.
Los pacientes con AP confluentes, tamaño normal y tronco pulmonar, normalmente con atresia valvular, son aptos para reparación compatible con Fallot mediante un parche transanular. Los pacientes con AP de tamaño normal pero sin tronco pulmonar deberían someterse a reparación con un conducto VD-AP. Los pacientes con AP confluentes pero hipoplásicas suelen requerir cortocircuito arterial o reconstrucción del TSVD, sin cierre del DSV, que quizá potencie el crecimiento de la AP, y luego una revisión en una fase posterior para la reparación con un conducto con válvula. Los pacientes con AP no confluentes y flujo sanguíneo pulmonar adecuado, pero no excesivo, en la infancia pueden sobrevivir hasta la edad adulta sin cirugía. Hay defensores de un abordaje estratificado de unifocalización para este difícil grupo de niños, que a la larga aspiran a la reparación de conducto264.
4.11.2Presentación clínica e historia naturalLa presentación clínica en la edad adulta en pacientes reparados es similar a la de aquellos con TdF (véase los apartados 4.10 y 4.14), mientras que los pacientes no reparados se presentan con disnea de esfuerzo, fatiga y cianosis crónica progresiva que se debe al menor flujo sanguíneo pulmonar secundario a la estenosis de las colaterales, estenosis de AP, mayor RVP o mayor presión ventricular diastólica final265. La cianosis puede llegar a afectar a múltiples órganos (véase el apartado 3.4.8) y con el tiempo puede aparecer una serie de complicaciones:
- •
La hemoptisis podría deberse a rotura de vasos colaterales normalmente pequeños o trombosis de la AP.
- •
La insuficiencia cardiaca crónica suele ser multifactorial y puede deberse a cianosis crónica, flujo sanguíneo pulmonar excesivo precoz, mayor RVP, disfunción del VD, IAo y otras causas.
- •
Podría haber dilatación progresiva de la aorta ascendente con mayor IAo y disección aórtica, aunque es una complicación muy poco común.
- •
La endocarditis puede ser especialmente de riesgo en pacientes con oca reserva cardiovascular o con cianosis importante.
- •
La arritmia y la MSC no son infrecuentes.
- •
HAP segmentaria46.
Véase el apartado 3.3 para conocer los principios generales.
- •
Hallazgos clínicos: podría haber cianosis profunda en pacientes no reparados, incluso con esfuerzo físico mínimo. Los soplos continuos en la parte posterior indican MAPCA. Los hallazgos del ECG incluyen desviación del eje a la derecha e HVD. La radiografía puede revelar un contorno cardiaco en forma de bota (arco pulmonar excavado) con vascularización pulmonar anómala y reducida alternando con algunas zonas de mayor vascularización a través de MAPCA grandes.
- •
Ecocardiografía: los hallazgos dependen del tipo de reparación (véase los apartados 4.10 y 4.14). En pacientes no reparados, podría faltar flujo directo desde el VD a la AP, con flujo continuo en múltiples puntos desde las MAPCA en el Doppler color. La ecocardiografía 3D puede ayudar a delinear la anatomía patológica y el tamaño y la función biventriculares. La ETE es útil en determinados pacientes para evaluar la anatomía de la válvula cuando sea difícil conseguir una buena imagen transtorácica o cuando se sospeche EI266.
- •
La RMC, la TCC y el cateterismo cardiaco son necesarios para determinar las fuentes del suministro sanguíneo pulmonar y el tamaño de las AP, así como para evaluar la HAP y las MAPCA. En pacientes reparados, la RMC se usa con los mismos fines que para los pacientes con TdF (evaluar la función y los volúmenes del VD, la IP, el tamaño, la forma y la expansión de las AP, el tamaño de la aorta ascendente y el cortocircuito residual [Qp:Qs]). La angiografía rotacional 3D, las imágenes de superposición 3D, la radiografía y la imagen de fusión por RMC pueden mejorar la precisión de la evaluación267.
Para el seguimiento y la intervención de pacientes con reparación tipo Fallot y parche transanular, véase el apartado 4.10; para pacientes con reparación mediante conducto VD-AP con válvula, véase el apartado 4.14.
Los pacientes con atresia pulmonar y DSV que sobreviven sin reparación hasta la edad adulta o con intervenciones paliativas previas quizá se beneficien de las intervenciones quirúrgicas modernas268,269. Podría ser el caso de pacientes con AP confluentes de tamaño normal, pero también aquellos con MAPCA anatómicamente grandes aptos para unifocalización que no han desarrollado enfermedad vascular pulmonar grave debido a la estenosis profiláctica. No obstante, muchos pacientes no reparados pueden no ser aptos para más cirugía, sobre todo por la complejidad de la vasculatura pulmonar. Es importante comprender que, aunque la cirugía cardiaca puede mejorar el estado clínico o pronóstico (que es puramente especulativo), también es una importante causa de mortalidad.
El cateterismo intervencionista puede incluir dilatación con balón/implante de stent en los vasos colaterales para mejorar el flujo sanguíneo pulmonar270. Por otro lado, los pacientes con hemoptisis grave pueden requerir implante de stent en los vasos colaterales rotos.
La supervivencia depende de la complejidad de las malformaciones pulmonares y los resultados de la reparación quirúrgica. La supervivencia de pacientes intervenidos con intención paliativa es significativamente menor, en torno al 60% a los 20 años de seguimiento. El trasplante de corazón-pulmón podría ser una opción para pacientes muy seleccionados.
4.11.5Recomendaciones sobre el seguimientoA los pacientes con atresia pulmonar y DSV se les debe hacer un seguimiento periódico en un centro especializado en CCA, al menos 1 vez al año. Para el tratamiento del deterioro de múltiples órganos secundario a la cianosis, véase el apartado 3.4.8.
Se debe considerar para tratamiento por objetivos de HAP a los pacientes con HAP segmentaria, aunque los datos son escasos (véase el apartado 3.4.3)271,272.
Merecen especial atención los síntomas como disnea, cianosis creciente, cambio en el soplo del cortocircuito, insuficiencia cardiaca o arritmias y requerirían una revisión y una evaluación para la intervención más precoces.
4.11.6Consideraciones adicionales- •
Ejercicio/deporte: se debe animar a los pacientes con excelente hemodinámica a practicar ejercicio con regularidad, aunque quizá deban evitar únicamente el ejercicio isométrico extremo. Aquellos con hemodinámicas subóptimas estarán más limitados funcionalmente. Los ejercicios de fuerza extremos o los deportes de contacto de competición se deben de evitar, aunque sí hay que fomentar la actividad física de baja intensidad, como caminar, nadar o incluso el ciclismo.
- •
Embarazo: el riesgo con el embarazo de las pacientes reparadas con buena hemodinámica y sin antecedentes de arritmias es bajo. El riesgo aumenta con hipoxemia, HAP, disfunción ventricular, síntomas de insuficiencia cardiaca y arritmias (véase el apartado 3.5.7). Puesto que la microdeleción del 22q11 es bastante común en este defecto, las pacientes deben pasar revisión antes del embarazo.
- •
Profilaxis de la EI: se ecomienda solo para pacientes en alto riesgo, incluidos todos los pacientes no reparados (véase el apartado 3.4.6).
La TGA se caracteriza por la concordancia AV y la discordancia ventriculoarterial: la aorta se origina del VD y la AP del VI. Si no hay lesiones cardiacas congénitas relevantes adicionales, se conoce como TGA simple. La TGA compleja tiene anomalías intracardiacas asociadas, como DSV (en hasta el 45% de los casos), OTSVI (25%) y CoA (5%). El resultado a largo plazo de la TGA compleja es peor que el de la TGA simple independientemente del tipo de reparación quirúrgica. La etiología de la TGA no se conoce y su patogénesis es controvertida. Existe la presentación familiar, pero es muy poco frecuente. Hay un predominio masculino de 2:1. La historia natural es extremadamente pobre y los pacientes que sobreviven hasta la edad adulta sin reparación quirúrgica son excepcionales. Las técnicas quirúrgicas han evolucionado en los últimos años: el switch auricular ha pasado a un procedimiento de cambio arterial, y los TGA complejos se suelen operar mediante una reparación tipo Rastelli.
En la figura 9 se muestra un esquema resumido de las técnicas quirúrgicas en la TGA y sus complicaciones a largo plazo.
 MSC: muerte súbita cardiaca;
MSC: muerte súbita cardiaca; Tratamiento de la transposición de las grandes arterias: complicaciones a largo plazo que atender en el seguimiento. AD: aurícula derecha; AI: aurícula izquierda; Ao: aorta; AV: auriculoventricular; AVP: aurícula venosa pulmonar; AVS: aurícula venosa sistémica; DNS: disfunción del nódulo sinusal; EP: estenosis pulmonar (rama arterial pulmonar/supravalvular); MSC: muerte súbita cardiaca; TSVI: tracto de salida del ventrículo izquierdo (subpulmonar); VCI: vena cava inferior; VCS: vena cava superior; VD: ventrículo derecho; VI: ventrículo izquierdo; VP: vena pulmonar; VT: válvula tricúspide.
Los adultos de más edad con TGA simple se habrán sometido a una intervención de Mustard o Senning con switch auricular. Las complicaciones más frecuentes son:
- •
Disfunción e insuficiencia del VD sistémico.
- •
IT progresiva secundaria (válvula AV sistémica).
- •
Bradicardia e incompetencia cronotrópica por pérdida del ritmo sinusal; la conducción AV suele estar intacta.
- •
Taquiarritmia supraventricular, típicamente flutter cavotricuspídeo dependiente del istmo, seguido de circuito de macrorreentrada relacionado con la incisión/cicatriz quirúrgica. Puede haber FA a edades más avanzadas. En general, las frecuencias cardiacas elevadas tienen mala tolerancia hemodinámica por la incapacidad para aumentar la precarga como consecuencia de los bafles auriculares (restrictivos). La bradicardia por disfunción del nodo sinusal puede provocar TA.
- •
Taquiarritmias ventriculares: TV polimórfica primara o FV por la mala función ventricular y por mecanismos relacionados con la insuficiencia cardiaca, o TV monomórfica debida a circuitos de reentrada relacionados con la cicatriz/incisión/parche en la TGA compleja; TV o FV secundarias, precedidas por taquicardia supraventricular (TSV) con conducción rápida e isquemia miocárdica consecutiva debida a un volumen sistólico muy bajo relacionado con la TSV.
- •
Estenosis del bafle, ya sea por obstrucción del bafle superior (el más frecuente) o del bafle inferior.
- •
Derrame o filtración del bafle, con cortocircuito I-D que causa aumento del flujo pulmonar o cortocircuito D-I en presencia de obstrucción distal del flujo, con cianosis o embolia paradójica.
- •
Obstrucción auricular venosa o de las venas pulmonares, más frecuentemente en el punto donde estas se conectan con la aurícula venosa pulmonar/AD.
- •
Puede producirse OTSVI por la protrusión del tabique interventricular hacia el VI subpulmonar de baja presión, frecuentemente asociado con el movimiento anterior sistólico de la válvula mitral.
- •
Puede aparecer HP, a veces décadas despues del procedimiento de switch auricular; suele ser poscapilar273, aunque también puede haber HAP.
- •
Muerte por insuficiencia cardiaca o muerte súbita, probablemente causada por una arritmia.
En las series de mayor tamaño con periodos de seguimiento de hasta 40 años, la supervivencia ha sido de un 60–75%274,275. La supervivencia sin complicaciones solo llega al 20%276,277. La capacidad de ejercicio está reducida debido al aumento inadecuado del gasto cardiaco: incompetencia cronotrópica, precarga reducida como consecuencia del estrechamiento de los bafles o bafles no permeables (inherente a la operación de switch auricular) y función reducida del VD.
4.11.10Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
La evaluación clínica debe incluir la búsqueda de signos de congestión venosa en las mitades superior e inferior del cuerpo. La cabeza y el cuello hinchados son un signo de obstrucción del bafle superior. La obstrucción del bafle inferior produce edema de las piernas, varices, hepatomegalia y cirrosis hepática. La estenosis, incluso la obstrucción completa, puede ser asintomática debido a una circulación eficaz de derivación proporcionada por la vena ácigos o hemiácigos.
El soplo sistólico compatible con eyección indica obstrucción del tracto de salida subpulmonar, y el soplo sistólico de tipo regurgitante, insuficiencia sistémica de la VT. Los hallazgos del ECG incluyen HVD y, con cierta frecuencia, ritmo de escape QRS estrecho, sin ondas P visibles.
- •
La ecocardiografía es la técnica diagnóstica de primera línea, ya que ofrece información sobre la función y los tamaños ventriculares sistémico y subpulmonar, la obstrucción del tracto de salida subpulmonar, la IT, las fugas o la obstrucción de los bafles auriculares y evaluación del retorno venoso pulmonar. Los signos de HP suelen ser sutiles (menor aplanamiento del septo interventricular en sístole y PA anormalmente ancha) y pueden ser difíciles de reconocer. La sospecha de HP debe inducir a cateterismo cardiaco diagnóstico para excluir/confirmar HP, ya que puede tener consecuencias en el tratamiento. La ecocardiografía de contraste está indicada si se sospecha derrame del bafle (presente en hasta el 50% de los pacientes asintomáticos no seleccionados) y es muy útil en la detección de estenosis del bafle278. La inyección de contraste en un brazo no suele detectar una filtración en el bafle inferior, que solo puede excluirse mediante inyección en una de las venas femorales. La ETE es útil para evaluar los bafles.
- •
La RMC permite evaluar la función del VD sistémico de modo más fiable y robusto que la ecocardiografía, así como la permeabilidad de los bafles auriculares. El tamaño de las grandes arterias se puede medir con fiabilidad; una AP anormalmente ancha o un VI subpulmonar grande pueden indicar HP. El cortocircuito relacionado con el derrame del bafle se puede cuantificar (Qp:Qs). Las filtraciones pequeñas del bafle que no producen un cortocircuito relevante son difíciles de detectar con RMC (la ecocardiografía de contraste es superior en estos casos). El realce tardío de gadolinio en el VD sistémico predice los resultados clínicos279.
- •
Es fundamental excluir y tratar una estenosis o filtración del bafle superior antes del implante de un DAI o MP o de la colocación de los cables de un dispositivo nuevo/adicional a través del bafle superior. Una alternativa a la RMC o la TCC para la evaluación del bafle superior puede ser la inyección de contraste en el brazo derecho y la fluoroscopia.
- •
La PECP es importante en el seguimiento longitudinal para evaluar de manera seriada la capacidad de ejercicio y la incompetencia cronotrópica. También puede desenmascarar el derrame del bafle (desaturación) que es asintomático en reposo.
- •
La monitorización Holter, el grabador de eventos y la prueba EF están indicadas para pacientes seleccionados cuando se sospechen bradicardia o taquiarritmias.
- •
El cateterismo cardiaco está indicado cuando la evaluación no invasiva sea inconcluyente o se deba evaluar la HAP (véase el apartado 3.3.5).
- •
Disfunción del VD sistémico: no hay datos que respalden la hipótesis de que los IECA, ARA–II, bloqueadores beta o antagonistas de la aldosterona (solos o combinados) mejoren los resultados clínicos280. Actualmente no se puede hacer una recomendación firme.
- •
Insuficiencia del VD sistémico: en caso de insuficiencia cardiaca manifiesta, los diuréticos alivian los síntomas. Aunque no se ha demostrado beneficio alguno del tratamiento médico convencional para la insuficiencia cardiaca en pacientes con VD sistémico, los pacientes más sintomáticos pueden beneficiarse de la prescripción de medicamentos «clásicos» para la insuficiencia cardiaca.
- •
Arritmia: los fármacos que reducen la frecuencia cardiaca deben usarse con precaución, ya que tras el switch auricular los pacientes son propensos a la bradicardia y la disfunción del nódulo sinusal.
- •
HP: es necesario comprender el mecanismo exacto de la HP antes de considerar el tratamiento médico. La HP poscapilar tardía tras la operación de switch auricular es la más frecuente, por lo que el tratamiento vasodilatador pulmonar está contraindicado, aunque la HP precapilar también puede estar presente. Por consiguiente, es fundamental una evaluación hemodinámica cuidadosa–
Pruebas EF, ablación, TRC y DAI.
Los principios generales, que también son válidos para los pacientes con switch arterial, se tratan en el apartado 3.4.232,37.
- •
Los estudios EF se ven complicados por el hecho de que las aurículas no suelen ser accesibles por catéter debido a la disposición de los bafles. El mecanismo principal de las arritmias supraventriculares es el flutter auricular del istmo cavotricuspídeo, que suele requerir punción del bafle para lograr el bloqueo del istmo. Como alternativa, la navegación magnética a distancia puede usarse para el acceso retrógrado a la aurícula venosa pulmonar. La ruta de acceso transaórtica retrógrada convencional no suele servir para conseguir el bloqueo del istmo en los adultos. Se recomienda ETE para guiar el catéter cuando está indicada la punción del bafle. La estimulación eléctrica programada no es útil para la estratificación del riesgo.
- •
Marcapasos: véase el apartado 3.4.2.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para la intervención quirúrgica y el cateterismo en la TGA tras la operación de switch auricular».
Recomendaciones para la intervención en la transposición de las grandes arterias tras switch auricular
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Indicaciones para la intervención quirúrgica | ||
| Para los pacientes con obstrucción venosa pulmonar sintomáticos se recomienda la reparación quirúrgica (casi nunca es posible el cateterismo intervencionista) | I | C |
| Para los pacientes con estenosis del bafle sintomáticos y no susceptibles de tratamiento percutáneo, se recomienda la reparación quirúrgica | I | C |
| Los pacientes sintomáticos con derrames del bafle no susceptibles de cierre percutáneo, se recomienda la reparación quirúrgica | I | C |
| Se debe considerar la reparación o sustitución de la válvula para los pacientes con insuficiencia grave de la válvula AV sistémica (tricúspide) sin disfunción ventricular relevante (FE > 40%) con independencia de los síntomas | IIa | C |
| No se recomienda la banda en la AP de pacientes adultos para crear un cambio septal o como entrenamiento del VI con posterior switch arterial | III | C |
| Indicaciones para el cateterismo intervencionista | ||
| Para los pacientes con estenosis del bafle sintomáticos, se recomienda el implante de stent cuando sea técnicamente posible | I | C |
| Para los pacientes sintomáticos con derrames del bafle y cianosis considerable en reposo o ejercicio o con firme sospecha de embolia paradójica, se recomienda el implante de stent (recubierto) o el cierre con dispositivo cando sea técnicamente posible | I | C |
| Para los pacientes con derrame del bafle y síntomas secundarios al cortocircuito I-D, se recomienda el implante de stent (recubierto) o el cierre con dispositivo cuando sea técnicamente posible | I | C |
| Para los pacientes asintomáticos con derrame del bafle y sobrecarga de volumen ventricular por cortocircuito I-D, se debe considerar el implante de stent (recubierto) o el cierre con dispositivo cuando sea técnicamente posible | IIa | C |
| Para los pacientes que requieren MP/DAI y cuando sea técnicamente posible, se debe considerar el cierre del derrame del bafle con implante de stent recubierto antes de la inserción de los cables transvenosos | IIa | C |
| Se puede considerar el implante de stent para los pacientes asintomáticos con estenosis del bafle cuando sea técnicamente posible | IIb | C |
AP: arteria pulmonar; AV: auriculoventricular; DAI: desfibrilador automático implantable; FE: fracción de eyección; I-D: izquierda-derecha; MP: marcapasos; VI: ventrículo izquierdo.
aClase de recomendación.
bNivel de evidencia.
Es muy probable que los adultos jóvenes con TGA simple se hayan sometido a una operación de switch arterial. Las complicaciones más frecuentes son:
- •
Dilatación de la raíz neoaórtica que produce IAo.
- •
EP supravalvular y estenosis de rama pulmonar (unilateral o bilateral), como consecuencia de la posición de la bifurcación pulmonar anterior a la aorta ascendente en la técnica de Lecompte y la dilatación de la raíz neoaórtica.
- •
La disfunción del VI y las arritmias ventriculares no son frecuentes, pero pueden ocurrir; ambos fenómenos pueden estar relacionados con problemas en las arterias coronarias281 que fueron reimplantadas en la neoaorta.
- •
Angulación aguda del arco aórtico, que puede producir obstrucción funcional e hipertensión.
La supervivencia hasta los 30 años es excelente (> 90% de supervivencia hospitalaria282) y la supervivencia sin complicaciones es aceptable (un 60-80%283–285). La gran mayoría de estos pacientes están asintomáticos. Como regla general, la capacidad de ejercicio se reduce levemente, pero puede ser normal. Se ha documentado que la incidencia de problemas tardíos relacionados con las arterias coronarias es muy baja286,287, lo que cuestiona la necesidad del cribado sistemático de las arterias coronarias.
4.11.13Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales. Puede haber hallazgos clínicos de IAo o EP.
- •
La ecocardiografía es la técnica diagnóstica clave, ya que ofrece información sobre la función del VI (general y regional), estenosis en los puntos arteriales anastomóticos, sobre todo EP, insuficiencia de la válvula neoaórtica, dimensión de la raíz neoaórtica y la aorta ascendente proximal y angulación aguda del arco aórtico. Se debe considerar la función del VD y evaluar las presiones sistólicas (velocidad de la regurgitación tricuspídea). Por su posición muy anterior y justo detrás del esternón, la visualización ecocardiográfica de la bifurcación y de las 2 ramas pulmonares rara vez es posible.
- •
La ecocardiografía de estrés puede desenmascarar la disfunción del VI y detectar isquemia miocárdica provocable.
- •
La RMC proporciona una evaluación cuantitativa más fiable de los volúmenes ventriculares, la FEVI y la dilatación o insuficiencia neoaórtica. Se puede visualizar el tronco pulmonar y las ramas pulmonares, así como su relación con la raíz neoaórtica (dilatada). Permite calcular la distribución del flujo entre el pulmón izquierdo y el derecho. La RMC de estrés es una técnica alternativa para evaluar la perfusión miocárdica y la posible afección coronaria cuando esté clínicamente indicado.
- •
La TCC es la técnica de elección para obtener imágenes no invasivas de las arterias coronarias, incluido el ostium, en caso de sospecha de estenosis. Debido a la baja incidencia de problemas relacionados con las arterias coronarias, no parece justificado el cribado sistemático con ninguna modalidad de imagen286,287.
- •
Las técnicas nucleares ya no se utilizan como modalidad de imagen de primera línea, pero pueden tener alguna utilidad cuando las otras técnicas no estén disponibles o produzcan resultados no concluyentes o contradictorios.
- •
El cateterismo cardiaco, incluida la angiografía coronaria, está indicado en caso de disfunción del VI y sospecha de isquemia miocárdica. También está indicado en caso de estenosis grave de las ramas pulmonares y evaluación no invasiva no concluyente o sospecha de HAP.
Las indicaciones para la intervención se resumen en la tabla «Recomendaciones para las intervenciones en la transposición de las grandes arterias tras operación de switch arterial».
Recomendaciones para la intervención en la transposición de las grandes arterias tras switch arterial
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Se recomienda el implante de stent o cirugía, dependiendo del sustrato, para la estenosis de arteria coronaria que causa isquemia | I | C |
| Se debe considerar la cirugía de la raíz neoaórtica cuando esta sea > 55mm, ofreciendo la estatura media del adulto (para la sustitución de la válvula aórtica en la IAo grave, véase la guía de valvulopatíasc) | IIa | C |
| Se debe considerar el implante de stent para la EP periférica, con independencia de los síntomas, si el estrechamiento del diámetro es > 50% y la presión sistólica del VD es > 50mmHg o hay anomalías en la perfusión pulmonar | IIa | C |
AP: arteria pulmonar; ESC: European Society of Cardiology; IAo: insuficiencia aórtica.
aClase de recomendación.
bNivel de evidencia.
cA la hora de aplicar la guía de la ESC/EACTS de 2017 sobre valvulopatías25 para la decisión de operar, hay que tener en cuenta que se trata de una reintervención y, por lo tanto, es técnicamente más difícil.
La mayoría de los adultos con TGA, DSV y EP (transposición compleja) se habrán sometido a una reparación de tipo Rastelli. El parche del DSV dirige la sangre desde el VI a la aorta, mientras que el VD está conectado a la AP con un conducto valvulado. Otras variantes de la técnica de Rastelli que comparten el mismo principio son la réparation à l’etage ventriculaire y la técnica de Nikaidoh.
Las principales complicaciones son:
- •
Estenosis o regurgitación del conducto valvulado entre el VD y la AP.
- •
OTSVI, es decir, obstrucción del flujo desde el VI a la aorta.
- •
DSV residual.
- •
IAo.
- •
Disfunción del VI.
- •
Arritmias, tanto ventriculares como supraventriculares.
- •
Endocarditis del conducto valvulado.
- •
Muerte, ya sea súbita (por arritmia) o por insuficiencia cardiaca.
Los pocos estudios de resultados a largo plazo indican una supervivencia a 20 años < 60% y una supervivencia a 20 años sin complicaciones de un 20–30%. La sustitución del conducto entre el VD y la AP es la indicación más frecuente para la reintervención. El alivio de la OTSVI es la segunda indicación más frecuente, seguida por el cierre de la DSV residual288. La endocarditis del conducto valvulado es relativamente común.
La reducción de la capacidad de ejercicio va de leve a importante. Nuevas reintervenciones quirúrgicas o percutáneas son el destino de la mayoría de los pacientes con una reparación tipo Rastelli.
4.11.15.2Estudio diagnósticoVéase el apartado 3.3. para conocer los principios generales.
Los hallazgos clínicos pueden indicar estenosis del conducto, DSV residual, IT, insuficiencia mitral o IAo.
• Se debe evaluar por ecocardiografía la conexión entre el VI posicionado posteriormente y la válvula aórtica posicionada anteriormente, por la TGA, la función de la válvula aórtica y los diámetros de la raíz aórtica. La anatomía y la función del conducto entre el VD y el tronco pulmonar deben visualizarse y evaluarse mediante Doppler. La presión del VD evaluada con Doppler a partir de la velocidad de la IT es especialmente importante, ya que la técnica Doppler a menudo sobrestima el gradiente de presión a través del conducto VD-PA.
–• La RMC permite una cuantificación más robusta de los volúmenes del VI y el VD, los diámetros aórticos y la FE. El conducto VD-AP, que suele ser difícil de visualizar por ecocardiografía, y las AP periféricas se pueden ver y medir fácilmente con la RMC. En presencia de DSV residual, es posible calcular el Qp:Qs.
–• El cateterismo cardiaco puede ser necesario en la evaluación hemodinámica de la estenosis del conducto. La angiografía puede ser útil en la evaluación del grado de estenosis y las estenosis de AP periféricas.
4.11.16Tratamiento quirúrgico/cateterismo intervencionistaVéase el apartado 4.14 para conocer las indicaciones sobre el tratamiento de la estenosis del conducto.
Si el cortocircuito I-D a través de un DSV residual causa síntomas o una considerable sobrecarga de volumen del lado izquierdo, se debe aplicar tratamiento quirúrgico (recomendación I C).
4.11.16.1Recomendaciones sobre el seguimiento (independientemente del tipo de reparación)Se debe atender a todos los pacientes con TGA en un centro especializado en CCA como mínimo 1 vez al año, con independencia del tipo de intervención. Se debe prestar especial atención a las cuestiones concretas ya descritas (véase los apartados 4.12.2.1, 4.12.3.1 y 4.12.4.1).
4.11.17Consideraciones adicionales (independientemente del tipo de reparación)• Ejercicio, embarazo y profilaxis de la EI: véase los apartados 3.4.6, 3.5.5 y 3.5.7.
4.12Transposición de las grandes arterias congénitamente corregida4.12.1Introducción y antecedentesLa TGAcc, o discordancia AV y ventriculoarterial, es poco común. Los ventrículos están invertidos, la aorta surge anteriormente del VD, normalmente en el lado izquierdo, y la AP surge posteriormente del VI, en el lado derecho. Pueden hallarse conexiones anómalas con doble discordancia en corazones con organización auricular normal o en espejo. La orientación cardiaca o eje base-vértice en dextrocardia (base del corazón orientada a la derecha) es frecuente, en torno al 20%. Las lesiones asociadas son comunes (80-90%), entre ellas DSV (70%), EP (40%) y VT sistémica displásica (p. ej., anomalía de tipo Ebstein).
Es frecuente que el nódulo AV (a veces, múltiples nódulos AV) y el haz de His estén en posición anómala y causen trastornos de la conducción. Es importante identificar el desplazamiento anterior y lateral del haz de His en los estudios EF y el cateterismo.
4.12.2Presentación clínica e historia naturalLa historia natural y la presentación clínica suelen estar determinadas por las malformaciones cardiacas asociadas. Los pacientes con lesiones asociadas normalmente alcanzan la edad adulta solo porque se operaron tiempo atrás —cierre del DSV, alivio de la EP o, más raramente, reparación o sustitución de la VT— o tienen una fisiología compensada. La TGAcc aislada raramente produce complicaciones antes de la edad adulta.
Las complicaciones tardías son:
- •
Disfunción e insuficiencia del VD sistémico.
- •
IT progresiva (válvula AV sistémica).
- •
OTSVI.
- •
Bloqueo AV completo (el 2% anual de pérdida de la conducción AV); es más habitual después de la reparación del DSV o sustitución de la VT y puede ocurrir durante el embarazo.
- •
TV (extremadamente infrecuentes).
La esperanza de vida está reducida: aproximadamente el 50% de los pacientes con lesiones asociadas alcanzan los 40 años; sin lesiones asociadas, el 50% de los pacientes siguen vivos a los 60 años. Los pacientes mueren por insuficiencia cardiaca congestiva o muerte súbita, probablemente por TV/FV, tengan insuficiencia cardiaca avanzada o no.
4.12.3Estudio diagnósticoVéase el apartado 3.3. para conocer los principios generales.
- •
Los hallazgos clínicos pueden incluir soplos o IT, DSV o EP.
- •
El ECG podría revelar intervalo PR prolongado o bloqueo cardiaco completo. Puesto que las ramas están invertidas, hay activación septal precoz D-I que podría causar ondas Q profundas en II, III, aVF y V1-V3. La inversión del avance precordial normal podría observarse como un patrón QR en V1 y rS en V6. Se halla síndrome de Wolf-Parkinson-White en un 2-4% de los pacientes.
- •
La radiografía de tórax puede revelar un borde izquierdo de la silueta cardiaca anormalmente recto por la posición anterior y hacia la izquierda de la aorta ascendente, dextrocardia (el 20%) o mesocardia (relativamente común).
- •
La ecocardiografía es la técnica diagnóstica clave para demostrar doble discordancia e identificar las anomalías asociadas (sobre todo malformación e insuficiencia de la VT compatible con la anomalía de Ebstein, DSV, OTSVI y EP). Se pueden evaluar cualitativamente la función del VD y el VI y la gravedad de la IT.
- •
La RMC ofrece la anatomía intracardiaca y de las grandes arterias y está indicada para la cuantificación de volúmenes, masa y FE ventriculares, sobre todo porque la evaluación ecocardiográfica de la función sistólica en los VD sistémicos es difícil y menos fiable.
- •
La monitorización Holter, el grabador de eventos y la prueba EF pueden estar indicados para la detección de arritmias y bloqueo AV progresivo y valorar el riesgo de MSC.
- •
El cateterismo cardiaco está indicado cuando la evaluación no invasiva sea no concluyente o sea necesario determinar la HAP (véase el apartado 3.4.5).
No hay datos que respalden la hipótesis de que los IECA, ARA–II, bloqueadores beta o antagonistas de la aldosterona, solos o combinados, mejoren los resultados clínicos280. No está recomendada la prescripción sistemática de estos medicamentos para prevenir la insuficiencia cardiaca o mejorar los resultados.
- •
En caso de insuficiencia cardiaca manifiesta, los diuréticos alivian los síntomas. Aunque no se ha demostrado beneficio del tratamiento médico convencional para la insuficiencia cardiaca en pacientes con VD sistémico, los pacientes más sintomáticos pueden beneficiarse de la prescripción de medicamentos clásicos o ARA–II para la insuficiencia cardiaca289. Una morfología de VD sistémico no es contraindicación para el DAI. La trabecularización gruesa del vértice del VD merece una consideración especial, ya que puede bloquear la cánula de entrada. Debe considerarse la miomectomía selectiva.
El cateterismo podría recomendarse para pacientes con estenosis de AP o del conducto que puede dilatarse o someterse a intervención con stent. No obstante, una OTSVI residual podría tener un efecto beneficioso en el VD sistémico dilatado (ventrículo subaórtico) y la insuficiencia de la válvula AV sistémica (tricúspide) por el cambio septal. La estimulación AV progresiva es el tratamiento estándar cuando se produce bloqueo completo. La fijación del cable ventricular en la débil pared del VI subpulmonar puede ser difícil y requiere experiencia. Algunos datos indican que el MP biventricular con un segundo cable ventricular a través del seno coronario por detrás del VD subaórtico puede ayudar a que la función sistólica del VD mejore más que el MP de VI solo290.
Recomendaciones para la intervención en la transposición de las grandes arterias congénitamente corregida
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Para los pacientes sintomáticos con IT grave y función del VD sistémico conservada o levemente deteriorada (FE > 40%), está indicada la sustitución de la VT | I | C |
| Para los pacientes asintomáticos con IT grave y dilatación progresiva del VD sistémico o función del VD sistémico levemente deteriorada (FE > 40%), se debe considerar la sustitución de la VT | IIa | C |
| Se debe considerar la estimulación biventricular en caso de bloqueo AV completo o necesidad de estimulación ventricular > 40% | IIa | C |
| Para los pacientes sintomáticos con IT grave y función del VD sistémico más que levemente reducida (FE ≤ 40%), se podría considerar la sustitución de la VT | IIb | C |
AV: auriculoventricular; FE: fracción de eyección; IT: insuficiencia tricuspídea; VD: ventrículo derecho; VT: válvula tricúspide.
aClase de recomendación.
bNivel de evidencia.
La IT, la disfunción del VI y el momento óptimo para implantar una VT o un DAI son los retos más importantes. A diferencia de la situación en el grupo de edad pediátrica, en el que el doble cambio es la opción terapéutica establecida para casos de insuficiencia del VD sistémico, este enfoque no suele tener éxito en los adultos.
La insuficiencia de la válvula AV sistémica (tricúspide) suele centrar la atención del tratamiento quirúrgico. La reparación no suele ser factible y, como regla general, la sustitución valvular es el tratamiento de elección. La presencia de una FEVD preoperatoria ≤ 40%, PAP > 50mmHg, FA y una clase funcional de la New York Heart Association (NYHA) III-IV se asocian con mortalidad tardía291.
4.12.6Recomendaciones sobre el seguimientoLos pacientes con TGAcc necesitan de por vida seguimiento en un centro especializado en CCA129 a intervalos anuales, sobre todo por los trastornos de la conducción y la disfunción ventricular sistémica y de la válvula AV sistémica. En caso de arritmias, véase el apartado 3.4.2.
4.12.7Consideraciones adicionales- •
Ejercicio/deporte: los pacientes con TGAcc sintomáticos y FEVD conservada deben evitar los deportes de competición. Los pacientes con lesiones asociadas relevantes o función del VD sistémico reducida deben limitarse a los deportes de baja intensidad.
- •
Embarazo: el riesgo depende del estado funcional, la función ventricular, la presencia de arritmias (sobre todo el bloqueo AV) y las lesiones asociadas (véase el apartado 3.5.7).
Los conductos establecen la continuidad entre el VD y la AP en defectos complejos cuando el tracto de salida nativo no es susceptible de reconstrucción, como en atresia pulmonar, tronco arterial común, TdF, síndrome de la válvula pulmonar ausente, intervención de Rastelli y operación de Ross.
Los tipos de conducto son con válvula (homoinjerto pulmonar o aórtico, válvulas bioprotésicas, conductos de venas yugulares bovinas [Contegra]) y sin válvula. No existe el conducto ideal. La poca durabilidad implica la reintervención precoz. Los factores predictivos de insuficiencia del conducto son el proceso de esterilización/conservación, un conducto más pequeño, el tipo de conducto, edad más joven al implante, la estenosis de AP y el diagnóstico de transposición269,292,293. Se ha comunicado un 32–40% de pacientes sin reintervención por insuficiencia del conducto a los 20 años269,292.
Entre las complicaciones, se incluyen sobrecrecimiento, obstrucción progresiva con o sin regurgitación, endocarditis y aneurismas o seudoaneurismas.
La presentación clínica puede ser con disnea de esfuerzo, palpitaciones, síncope y MSC.
4.13.2Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos pueden ser vibración precordial, onda A de las venas yugulares prominente y soplo sistólico. La radiografía puede revelar calcificación del conducto.
- •
La ecocardiografía es la herramienta diagnóstica de primera línea, ya que muestra el tamaño y la función de ambos ventrículos, la IP, la IT y las lesiones asociadas. Los gradientes a través del conducto pueden ser difíciles de medir, además de poco fiables. Para evaluar la estenosis del conducto, se debe emplear la presión del VD obtenida a partir de la velocidad de la IT.
- •
La RMC se usa para cuantificar la estenosis o regurgitación del conducto y la masa y el volumen del VD y evaluar las AP.
- •
La RMC y la TCC pueden ser necesarias para definir la anatomía coronaria y la relación entre el VD/conducto y la capa interna del esternón.
- •
El cateterismo cardiaco con evaluación hemodinámica siempre es necesario si se considera intervenir. La angiografía ofrece información sobre el grado de estenosis, la estenosis de AP periférica y la anatomía coronaria (anomalías/curso anómalo).
La dilatación con balón y el implante de stent son seguros y podrían prolongar la vida útil de los dispositivos que fallan294,295. El IPVP se ha convertido en el tratamiento de elección para las válvulas disfuncionales, siempre que sea técnicamente factible. Las contraindicaciones actuales a la IPVP incluyen venas centrales ocluidas, infección activa, tracto de salida de morfología desfavorable y anatomía coronaria desfavorable (compresión por el implante extendido). Se prefiere la cirugía cuando se consideran más intervenciones (anuloplastia tricuspídea). Los datos longitudinales son más importantes que los indicadores únicos para elegir el momento de la reintervención.
Recomendaciones para la intervención en pacientes con conductos del ventrículo derecho a la arteria pulmonar
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Los pacientes sintomáticos con presión sistólica del VD > 60mmHg (puede ser inferior en caso de flujo reducido) o IP gravec deben someterse a intervención, preferiblemente percutánea (IPVP) si es anatómicamente factible | I | C |
| Para los pacientes asintomáticos con OTSVD grave o IP grave, se debe considerar la intervención, preferiblemente percutánea (IPVP) si es anatómicamente factible, cuando se cumpla al menos 1 de los siguientes criterios:• Reducción objetiva de la capacidad de ejercicio (PECP)• Dilatación progresiva del VD a VTSVDi ≥ 80 ml/m2 o VTDVDi ≥ 160 ml/m2 y progresión de la IT a ≥ moderada• Disfunción sistólica del VD progresiva• Presión sistólica del VD > 80 mmHg | IIa | C |
IP: insuficiencia pulmonar; IPVP: implante percutáneo de la válvula pulmonar; IT: insuficiencia tricuspídea; OTSVD: obstrucción del tracto de salida del ventrículo derecho; PECP: prueba de ejercicio cardiopulmonar; RMC: resonancia magnética cardiovascular; VD: ventrículo derecho; VTDVDi: volumen telediastólico del ventrículo derecho indexado; VTSVDi: volumen telesistólico del ventrículo derecho indexado.
aClase de recomendación.
bNivel de evidencia.
cFracción regurgitante por RMC > 30-40%.
Se recomienda un seguimiento regular en un centro especializado en CCA, al menos cada 12 meses. Debe prestarse especial atención a la capacidad de ejercicio (PECP), la presión sistólica del VD (gradiente del conducto), la función del VD, la IT y las arritmias.
4.13.4Consideraciones adicionales- •
Ejercicio/deporte: los pacientes asintomáticos con obstrucción leve no precisan restricciones. Los pacientes con alto riesgo y presión del VD elevada deben limitarse a las actividades/deportes de baja intensidad y evitar el ejercicio isométrico. Los demás deberían limitarse conforme a sus síntomas.
- •
Embarazo: los riesgos maternos y fetales se deben al defecto cardiaco congénito de base y la gravedad de la OTSVD, la arritmia y la insuficiencia cardiaca (véase el apartado 3.5.7).
- •
Profilaxis de la EI: se recomienda para todos los pacientes (véase el apartado 3.4.6).
Este apartado aborda el corazón univentricular (CUV) no intervenido o intervenido con intención paliativa. Para los pacientes tras operación de Fontan, véase el apartado 4.16.
4.14.1Introducción y antecedentesEl término «corazón univentricular» resume una variedad de malformaciones en las que falta el VD o el VI o, cuando no, son hipoplásicos y, por lo tanto, no susceptibles de reparación biventricular, como:
- •
Atresia tricuspídea.
- •
Variantes del síndrome del corazón derecho hipoplásico (p. ej., atresia pulmonar con variantes intactas del septo ventricular).
- •
Variantes del síndrome del corazón izquierdo hipoplásico, incluida la atresia mitral.
- •
VI de doble entrada.
- •
VD de doble entrada.
- •
Formas extremas de defectos del septo AV completos descompensados.
- •
Ventrículo único con morfología indefinida.
Estas malformaciones siempre están relacionadas con lesiones intracardiacas o extracardiacas adicionales, como:
- •
CIA, DSV, DSAV y DAP.
- •
EA (valvular, subvalvular).
- •
Anomalías del arco aórtico: hipoplasia, interrupción y coartación.
- •
EP (valvular, subvalvular) y atresia pulmonar.
- •
Anomalías de la AP: estenosis periférica, hipoplasia y ausencia unilateral.
- •
Conexiones discordantes y ectopia de las grandes arterias.
- •
Estenosis de la válvula AV, insuficiencia y cabalgamiento.
- •
Isomerismo de la AI o la AD y conexiones venosas sistémicas o pulmonares anómalas.
- •
VCS izquierda, vena innominada ausente, VCS derecha ausente y VCI infrahepática ausente con continuidad de la ácigos o hemiácigos.
- •
Arterias colaterales aortopulmonares.
- •
Poliesplenia o asplenia.
La descripción anatómica detallada va más allá del ámbito de esta guía y puede encontrarse en los libros de texto. Debido a la falta de datos, las recomendaciones son, principalmente, fruto del consenso de expertos296–300. Al presentarse ya adultos, la gran mayoría de los pacientes con estas afecciones se habrán sometido a paliación previa con algún tipo de derivación sistémica-PA, conexión cavopulmonar (Glenn) o ahora, preferiblemente, operación de Fontan o una de sus modificaciones; esta se trata en el apartado 4.16.
Se pueden identificar 2 situaciones hemodinámicas diferentes:
- •
Sin restricción anatómica del flujo pulmonar: si la circulación pulmonar sigue igual, es decir, sin cirugía, muchos pacientes mueren en la infancia por insuficiencia cardiaca incurable. Los que sobrevivan a este periodo contraerán enfermedad vascular pulmonar grave, que es un determinante fundamental del resultado a largo plazo. Muchos se han sometido a implante de banda pulmonar para restringir el flujo sanguíneo pulmonar en los primeros años de la infancia. Una banda eficaz protege contra la enfermedad vascular pulmonar al mismo tiempo que permite el paso de flujo sanguíneo pulmonar suficiente para limitar el grado de cianosis. Una banda poco ajustada causa sobreflujo pulmonar y enfermedad vascular pulmonar a pesar de la banda. Si está demasiado ajustada, el flujo sanguíneo pulmonar queda muy limitado y origina cianosis grave.
- •
Con obstrucción del flujo sanguíneo pulmonar (frecuentemente EP valvular o subvalvular o atresia): a veces, la obstrucción es tal que la circulación pulmonar es adecuada, no excesiva, lo que evita el desarrollo de hipertensión pulmonar, ni demasiado restringida, sin cianosis extrema. Estas situaciones equilibradas son la excepción, pero permiten la supervivencia hasta la edad adulta sin cirugía. La mayoría de los pacientes tienen flujo sanguíneo pulmonar muy restringido y requieren operación de cortocircuito de sistémica a AP en la infancia, muy comúnmente de Blalock-Taussig (subclavia a AP) y pocas veces de Waterston o Potts (aorta ascendente o descendente a AP respectivamente). Si el circuito de sistémica a AP es demasiado grande, el sobreflujo pulmonar resulta en enfermedad vascular en la edad adulta. Si el cortocircuito es demasiado pequeño, los pacientes serán extremadamente cianóticos. Después de la infancia, la anastomosis entre VCS y AP es una posibilidad: la clásica anastomosis de Glenn a la AP derecha (histórica) o una anastomosis terminolateral con la AP, lo que crea anastomosis cavopulmonar bidireccional. Un cortocircuito adecuado dará lugar a una situación equilibrada.
Dependiendo del alcance del flujo sanguíneo pulmonar, la presencia o ausencia de enfermedad vascular pulmonar y la función ventricular, los pacientes pueden presentarse con diferentes grados de cianosis e insuficiencia cardiaca congestiva. En general, la capacidad de ejercicio se reduce considerablemente, con excepciones; puede haber bloqueo AV completo, arritmias (supraventriculares, pero también ventriculares; la MSC es común), accidentes cerebrovasculares, abscesos cerebrales y tromboembolias. La endocarditis es relativamente común en esta población. Para más detalles, véase el apartado 3.4.8.
La cianosis es típica de los pacientes con CUV sin operación de Fontan. Normalmente, la saturación de oxígeno arterial oscila entre el 75 y el 85%, pero en casos excepcionales con las ideales circulaciones equilibradas, podría alcanzar valores > 90%.
Los pacientes pueden presentarse con obstrucción progresiva hacia la aorta. Esto origina hipertrofia ventricular y, finalmente, gasto cardiaco reducido. La obstrucción progresiva hacia la AP causa cianosis progresiva. En los pacientes de Glenn, el empeoramiento de la cianosis podría deberse también al desarrollo de malformaciones AV pulmonares o colaterales de VCI a VCS.
El CUV debe dar cabida al retorno venoso sistémico y pulmonar. Su sobrecarga de volumen crónica incrementa la probabilidad de insuficiencia ventricular relativamente pronto en la vida. Podría aparecer insuficiencia de la válvula AV o avanzar si ya se había iniciado. La capacidad de ejercicio ya reducida se deteriorará aún más. Al final, podría desarrollarse insuficiencia cardiaca manifiesta, además de cianosis.
En casos poco frecuentes, con situación hemodinámica bien equilibrada, no se desarrolla disfunción ventricular, y se ha comprobado supervivencia hasta la quinta, la sexta e incluso la séptima década de vida.
4.14.3Estudio diagnósticoVéase el apartado 3.3 para conocer los principios generales.
Los hallazgos clínicos incluyen cianosis central, acropaquia en los dedos de manos y pies y un tórax a menudo asimétrico con movimiento precordial del lado del pecho que aloja el corazón. La escoliosis es una complicación común. El segundo ruido cardiaco suele ser único, pero el resto de la auscultación depende de las anomalías asociadas. El ECG quizá revele trastornos de ritmo o conducción. Tanto la taquicardia auricular reentrante con bloqueo 2:1 como la taquicardia solamente moderada podrían pasarse por alto fácilmente.
• La ETT es la técnica diagnóstica clave, ya que ofrece información sobre la anatomía y monitoriza la función cardiaca durante el seguimiento. El enfoque segmentado es necesario en los exámenes ecocardiográficos, ya que los CUV siempre son complejos y pueden aparecer con un amplio abanico de anomalías de localización, orientación y conexiones. Lo fundamental en el diagnóstico de CUV es:
- -
Situs abdominal y situs auricular.
- -
La posición del corazón en el pecho y la posición del vértice.
- -
Conexión venoauricular, AV y ventriculoarterial.
- -
Hay que obtener información morfológica y hemodinámica de todo el corazón.
- -
Hay que evaluar la anatomía exacta de la conexión ventriculoarterial y su estado funcional, con especial atención a la obstrucción hacia la aorta o el lecho vascular pulmonar.
- -
Debe evaluarse la función de la válvula AV y prestar especial atención a la insuficiencia.
- -
Función/hipertrofia ventricular.
- -
Tipo, tamaño, número y ubicación de CIA/DSV.
- -
Aorta ascendente, arco aórtico y aorta descendente; detectar/excluir coartación.
- -
AP: tronco, ramas y fuentes de suministro del flujo sanguíneo pulmonar comunes.
- -
Visualización de los cortocircuitos, de Blalock-Taussig, de Waterston, etc.
- •
La ETE podría estar indicada en casos de imágenes de ETT inadecuadas.
- •
La RMC es la modalidad de imagen de elección para la anatomía extracardiaca, incluidas las conexiones venoauriculares y ventriculoarteriales; la TCC es una alternativa. También se puede obtener información morfológica detallada de la anatomía intracardiaca. La RMC también es el método de elección para cuantificar los volúmenes ventriculares, la FE y la distribución relativa del flujo sanguíneo en los pulmones izquierdo y derecho.
- •
El cateterismo cardiaco es necesario cuando se considera intervenir para la evaluación hemodinámica, en concreto de PAP y gradiente transpulmonar; la RVP suele ser difícil de evaluar en esta situación. Es obligatorio cuando se evalúa a los pacientes para operación de Fontan. La evaluación de cortocircuitos sistémicos a AP o de Glenn y sus secuelas (estenosis de las ramas pulmonares) y otras anomalías vasculares (vasos colaterales arteriovenosos, fístulas, etc.) podría requerir cateterización.
Consideraciones y recomendaciones especiales para la intervención en el corazón univentricular
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Se recomienda que los adultos con CUV no operado o paliado se sometan a una evaluación cuidadosa en centros especializados que incluya técnicas multimodales de imagen y a una evaluación diagnóstica invasiva para decidir si pueden beneficiarse de un procedimiento quirúrgico o intervencionista | I | C |
| Solo se debe considerar candidatos a circulación de Fontan a los pacientes cianóticos sintomáticos bien seleccionados tras una evaluación cuidadosa (resistencia vascular pulmonar baja, función adecuada de las válvulas AV y función ventricular conservada) | IIa | C |
| Se debe considerar la colocación de una banda en AP o el ajuste de una ya colocada en los pacientes con flujo sanguíneo pulmonar aumentado, poco probable en la edad adulta | IIa | C |
| Para los pacientes con cianosis grave y flujo sanguíneo pulmonar reducido sin RVP elevada, se debe considerar un cortocircuito de Glenn bidireccional | IIa | C |
| Para los pacientes con cianosis grave y flujo sanguíneo pulmonar reducido no aptos para cortocircuito de Glenn, se puede considerar un cortocircuito sistémico a AP | IIb | C |
| Para los pacientes con mal estado clínico, se debe considerar el trasplante cardiaco y el trasplante de corazón-pulmón cuando no haya opción quirúrgica convencional | IIa | C |
AP: arteria pulmonar; AV: auriculoventricular; CUV: corazón univentricular; PAP: presión arterial pulmonar; RVP: resistencia vascular pulmonar.
aClase de recomendación.
bNivel de evidencia.
Son discutibles las intervenciones como la valvulotomía pulmonar para aumentar el flujo sanguíneo pulmonar en casos de EP grave.
Si la situación clínica es estable, el riesgo, frecuentemente alto, de cualquier tipo de intervención quirúrgica debe sopesarse muy cuidadosamente con el posible beneficio.
La operación de Fontan solo puede considerarse para pacientes muy bien seleccionados (véase el apartado 4.16). Para pacientes con cianosis grave y flujo sanguíneo pulmonar reducido sin RVP elevada, el cortocircuito de Glenn bidireccional (de VCS a AP) puede ser una opción. Si el cortocircuito de sistémica a pulmonar (p. ej., fístula arteriovenosa axilar o cortocircuito de sistémica a AP) es la única opción porque el cortocircuito de Glenn bidireccional no es suficiente o la PAP no es lo bastante baja para este cortocircuito, el beneficio del mayor flujo sanguíneo pulmonar debe sopesarse con la mayor carga de volumen al ventrículo sistémico.
En caso de trasplante, tanto las esternotomías/toracotomías previas como las colaterales aortopulmonares y la naturaleza multisistémica de la CC cianótica son retos técnicos y médicos que limitan el resultado.
4.14.4Tratamiento conservadorPara el tratamiento hematológico y la utilidad del tratamiento dirigido en la enfermedad vascular pulmonar, véase los apartados 3.4.3 y 3.4.8 respectivamente.
4.14.5Recomendaciones sobre el seguimientoEs necesaria la evaluación regular en centros especializados en CCA.
La frecuencia es individualizada, pero anual como mínimo, con reconocimiento físico, medición de la saturación de oxígeno, controles de laboratorio (índices hematológicos, estado del hierro, función renal, etc.), ECG, radiografía y ecocardiografía (véase también el apartado 3.4.8).
Son necesarias una RMC y una prueba de ejercicio al menos 1 vez en la edad adulta, y en posteriores ocasiones a intervalos según los hallazgos basales.
4.14.6Consideraciones adicionales- •
Ejercicio/deporte: como norma, los pacientes no tienen aumentado el riesgo de muerte durante el ejercicio, pero sí una capacidad de ejercicio considerablemente reducida. Los deportes recreativos pueden considerarse a un nivel limitado según los síntomas.
- •
Embarazo: está contraindicado para pacientes con flujo sanguíneo pulmonar reducido grave o enfermedad vascular pulmonar grave o si la función ventricular es mala. La cianosis supone un riesgo considerable para el feto, y el nacimiento vivo es poco probable (< 12%) si la saturación de oxígeno es ≤ 85%106 (véase el apartado 3.5.7).
- •
Para la anticoncepción, deben evitarse las píldoras anticonceptivas orales combinadas por el riesgo trombogénico y tromboembólico. Las píldoras de progestágeno solamente y los dispositivos intrauterinos o los sistemas de implante liberadores de progestágeno ofrecen una anticoncepción segura con menos riesgo cardiovascular.
- •
Profilaxis de la EI: está indicada para todos los pacientes (véase el apartado 3.4.6).
La operación de Fontan se introdujo en 1968 y se ha convertido en el tratamiento definitivo para pacientes aptos con un abanico de malformaciones cardiacas caracterizadas por un único ventrículo funcional (véase el apartado 4.15). La cirugía consiste en la separación de los retornos venosos sistémico y pulmonar sin un ventrículo subpulmonar, restaurándolos para que estén «en serie». Desde su introducción, la intervención original, diseñada para optimizar el retorno venoso sistémico a las arterias pulmonares, ha sufrido varias modificaciones. En la actualidad, la conexión cavopulmonar total ha reemplazado a la conexión auriculopulmonar (apéndice auricular derecho a AP), con un conducto intracardiaco o extracardiaco entre la VCI y la AP, junto con anastomosis de VCS a AP (Glenn bidireccional)301. Esta circulación suele establecerse en 2 fases. Los adultos con hipoplasia del corazón izquierdo siguen siendo un grupo de pacientes pequeño aunque en expansión. Los primeros resultados en la edad adulta describen una prevalencia considerable de eventos cardiovasculares adversos mayores y una mayor propensión a complicaciones que los pacientes con Fontan general, lo que justifica un seguimiento y una evaluación más estrechos302.
Tanto la historia natural como los resultados de otras paliaciones de CUV son malos, por lo que la operación de Fontan suele realizarse en todos los pacientes con hemodinámica apta. Se comprende ahora que la mortalidad operatoria y el posterior resultado dependen de lo apropiado de la circulación y la adherencia a los criterios definidos. La selección estricta facilita mejores resultados precoces y tardíos, con una mortalidad operatoria < 5% en series modernas, e incluye RVP y PAP bajas (media < 15mmHg), función ventricular conservada, tamaño adecuado de AP, insuficiencia de la válvula AV no relevante y ritmo normal. Algunos centros han realizado fenestración en casos seleccionados o en todos los casos para permitir la derivación de la sangre desoxigenada a la circulación sistémica a la aurícula, con el objetivo de mejorar el gasto cardiaco a expensas de la cianosis303. Debido a los limitados resultados a largo plazo, la operación de Fontan no siempre es el método paliativo de elección.
4.15.2Presentación clínica e historia naturalLa falta de un ventrículo subpulmonar resulta en hipertensión venosa sistémica crónica, hemodinámica pulmonar notablemente alterada y un ventrículo «privado de precarga». Han surgido algunas complicaciones importantes durante el seguimiento a largo plazo. Aunque la supervivencia a los 10 años quizá se acerque al 90%, debe comprenderse que se observa inevitablemente una disminución prematura del rendimiento cardiovascular, con una supervivencia reducida, en los mejores pacientes de la operación de Fontan304. Hay cuestiones hemodinámicas importantes que contribuyen al fracaso tardío de la operación de Fontan: disminución gradual de la función ventricular sistémica, insuficiencia de la válvula AV, aumento en la RVP, agrandamiento auricular, obstrucción venosa pulmonar, DSV subaórtico progresivamente restrictivo y las consecuencias de la hipertensión venosa sistémica crónica, como congestión y disfunción hepáticas305. Otras complicaciones incluyen la formación de trombos auriculares y de AP, el desarrollo de malformaciones arteriovenosas pulmonares, la conexión sistémica de la arteria a la vena pulmonar o de la arteria a la arteria pulmonar, y de colaterales venosas sistémicas a pulmonares.
Tras la operación de Fontan, la mayoría de los pacientes responden bien durante la infancia y la adolescencia, aunque la capacidad de ejercicio medida objetivamente se reduce. No obstante, las complicaciones clínicas podrían aparecer después, con un descenso gradual del rendimiento durante el ejercicio e insuficiencia cardiaca, cianosis, insuficiencia venosa crónica y aparición de arritmias importantes, sobre todo en pacientes con operación de Fontan clásica306. Diez años después de la operación de Fontan, un 20% de los pacientes tienen TSV, que en general incluyen TRIA y flutter auricular, pero también FA y taquicardia auricular focal307. La bradicardia por disfunción del nódulo sinusal puede facilitar la aparición de TA. La incidencia de TA es menor tras la conexión cavopulmonar total que tras la conexión auriculopulmonar, y menor en el conducto extracardiaco que en el intracardiaco308. Las taquiarritmias auriculares con conducción rápida se asocian con MSC.
El espectro de hepatopatía asociada con Fontan es amplio e incluye congestión hepática y fibrosis grave, con signos de hipertensión portal y nódulos hipervasculares, así como carcinoma hepatocelular309.
La enteropatía pierdeproteínas es una complicación rara pero importante que resulta en edema periférico, derrames pleurales y ascitis. Puede diagnosticarse por documentarse escasez de albúmina en suero y aumento de la concentración de α1-antitripsina en heces310. Se ha asociado con un pronóstico muy malo (supervivencia a los 5 años < 50%), aunque un estudio reciente ha documentado una supervivencia a los 5 años del 88%. No obstante, su tratamiento sigue siendo un desafío médico311. La bronquitis plástica y la disfunción del sistema linfático pueden complicar el pronóstico aún más.
Véase una revisión extensa publicada recientemente para conocer más detalles312.
4.15.3Estudio diagnósticoVéase el apartado 3.3. para conocer los principios generales.
Los hallazgos clínicos incluyen distensión venosa yugular, comúnmente leve y no pulsátil. Sin embargo, la distensión venosa yugular relevante y la hepatomegalia plantean la sospecha de obstrucción de Fontan o insuficiencia ventricular. El ECG suele revelar ritmo de unión o arritmias auriculares. El derrame pleural en la radiografía hace sospechar la enteropatía pierdeproteínas.
- •
La ecocardiografía es la herramienta diagnóstica de primera línea, ya que ofrece información sobre la función ventricular y valvular. Para reflejar en imágenes la vía de Fontan, suele requerirse ETE u otras modalidades de imagen.
- •
Los análisis sanguíneos anuales incluyen hematología, concentración de albúmina en suero y función hepática y renal. Cuando se sospeche enteropatía pierdeproteínas, es necesario calcular el aclaramiento de α1-antitripsina.
- •
La RMC y la TCC son útiles para evaluar la vía de Fontan y las venas colaterales y pulmonares (p. ej., obstrucción de vena pulmonar por AD agrandada) y detectar trombos. LTCC requiere experiencia para mitigar el artefacto de transmisión y el diagnóstico falso positivo de trombo. La RMC se realiza con regularidad para los volúmenes ventriculares, la permeabilidad y los flujos de la vía de Fontan, para evaluar la insuficiencia de la válvula AV, la obstrucción subaórtica, la fibrosis miocárdica y la detección de trombos.
- •
Como la disfunción hepática, se debe reconocer la cirrosis y el carcinoma hepatocelular como complicaciones típicas; se debe hacer con regularidad estudios por imagen hepática (ecografía, TC, RM) y análisis de laboratorio.
- •
El cateterismo cardiaco debe realizase en un umbral bajo en casos de edema inexplicable, deterioro de la capacidad de ejercicio, arritmia de nueva aparición, cianosis y hemoptisis. Proporciona información sobre la función ventricular y valvular, hemodinámica (incluida la RVP) y obstrucción de Fontan y conexiones vasculares anómalas (véase el apartado 4.16.2). La integración con la RMC para los flujos (gasto cardiaco) permite una cuantificación más precisa de la RVP.
- •
Anticoagulación: la estasis sanguínea de la AD y la coagulación alterada pueden predisponer a la trombosis. El potencial de sufrir una embolia pulmonar subclínica y recurrente que cause un aumento de la RVP y una embolia sistémica ha propiciado la recomendación de administrar anticoagulación de por vida. No obstante, no hay pruebas de su beneficio y la práctica varía entre centros. La anticoagulación está recomendada, sin duda, en presencia de trombo auricular, arritmias auriculares o eventos tromboembólicos. Aunque los NACO se han demostrado seguros en pacientes seleccionados con circulación de Fontan79, aún no se dispone de datos prospectivos sólidos, por lo que estos fármacos no están recomendados como tratamiento estándar.
- •
Tratamiento antiarrítmico: la pérdida de ritmo sinusal podría precipitar un deterioro hemodinámico grave, y la arritmia sostenida se debería considerar urgencia médica. La cardioversión eléctrica es el pilar del tratamiento, ya que el tratamiento farmacológico no suele ser eficaz. La amiodarona podría ser eficaz en la prevención de recurrencias, pero tiene muchos efectos secundarios a largo plazo. El sotalol puede ser una alternativa. El umbral para la ablación por radiofrecuencia debería ser bajo, aunque son arritmias difíciles de tratar en el laboratorio de cateterismo313. El MP antitaquicardia auricular podría ser de ayuda. En caso de que se requiera un MP AV, será necesario aplicar un abordaje epicárdico. La aparición de arritmias puede dar lugar a la evaluación hemodinámica. Se debe considerar un enfoque proactivo de evaluación EF y ablación cuando esté indicada, incluida la conversión de Fontan con cirugía por arritmia. Se puede considerar el implante de un DAI en pacientes seleccionados (véase el apartado 3.4.2).
- •
Tratamiento médico para la enteropatía pierdeproteínas: sigue siendo un reto, y se han propuesto varios tratamientos tras exclusión de complicaciones hemodinámicas, como la restricción de sal, una dieta alta en proteínas, diuréticos, IECA (aunque pueden tolerarse muy mal), esteroides, infusión de albúmina, heparina subcutánea crónica, creación de fenestración (intervención por cateterismo) y, finalmente, consideración de trasplante.
- •
Vasodilatadores pulmonares: se puede considerar el tratamiento con ARE e inhibidores de la PDE-5 para pacientes seleccionados con aumento de la presión/resistencia pulmonar cuando no haya aumento de la presión ventricular telediastólica. En la actualidad hay muy pocos datos sobre el empleo sistemático de estas medicaciones en pacientes con Fontan. Un estudio clínico aleatorizado con bosentán (un ARE) ha demostrado una mejora significativa en la capacidad de esfuerzo cardiopulmonar en 75 adultos con fisiología de Fontan314.
Se debería considerar para cirugía a los pacientes con «fracaso de Fontan» (con combinación de arritmia intratable, dilatación auricular derecha, empeoramiento de la insuficiencia de la válvula AV, deterioro de la función ventricular o trombo auricular)315. Si bien los pacientes con función ventricular sistémica deficiente pueden beneficiarse del trasplante cardiaco (realizado en centros con experiencia), los pacientes con función ventricular sistémica conservada, arritmia auricular y dinámica de flujo alterada en la vía de Fontan pueden beneficiarse de la conversión de una conexión auriculopulmonar para que sea una conexión cavopulmonar total «energéticamente eficaz», junto con crioablación316. Esta intervención ha proporcionado buenos resultados precoces en un contexto de gran experiencia, aunque se ha asociado con mortalidad quirúrgica y morbilidad en curso, y la mayoría de los casos necesitan tanto tratamiento farmacológico como implante de MP317. Si se realiza tarde, la conversión podría tener menos probabilidades de arrojar un buen resultado, y se necesitaría un trasplante cardiaco. No obstante, el mejor momento para una conversión sigue siendo una cuestión incierta. En pacientes adultos seleccionados, quizá sea apropiado considerar el cierre con dispositivo de una fenestración si hay cianosis relevante, aunque también podría empeorar la enfermedad del paciente. El cateterismo también podría ser necesario en caso de obstrucción del flujo o conexiones vasculares anómalas.
Consideraciones y recomendaciones especiales para la intervención tras la operación de Fontan
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| La arritmia auricular sostenida con conducción AV rápida es una urgencia médica y debe tratarse de inmediato con cardioversión eléctrica | I | C |
| La anticoagulación está indicada cuando haya trombo auricular, arritmias auriculares y episodios tromboembólicos o antecedentes | I | C |
| Se recomienda asesorar contra el embarazo a las mujeres con circulación de Fontan y cualquier complicación | I | C |
| Se recomienda el cateterismo cardiaco a un umbral bajo en casos de edema inexplicable, deterioro de la capacidad de ejercicio, arritmia de nueva aparición, cianosis y hemoptisis | I | C |
| Para los pacientes con arritmias, se debe considerar un enfoque proactivo de evaluación electrofisiológica y ablación (cuando sea apropiado) | IIa | C |
| Se debe considerar la obtención periódica de imágenes hepáticas (ecografía, tomografía computarizada, resonancia magnética) | IIa | C |
| Se pueden considerar los antagonistas del receptor de la endotelina y los inhibidores de la fosfodiesterasa-5 para pacientes seleccionados con presión/resistencia pulmonar elevada en ausencia de presión telediastólica ventricular elevada | IIb | C |
| Se puede considerar el cierre con dispositivo de una fenestración en pacientes seleccionados con cianosis significativa, pero requiere una evaluación cuidadosa antes de la intervención para excluir la inducción de un aumento de la presión venosa sistémica o una caída del gasto cardiaco | IIb | C |
AV: auriculoventricular.
aClase de recomendación.
bNivel de evidencia.
Como resultado de la complejidad de todas estas cuestiones, el cuidado de los pacientes con Fontan es uno de los mayores retos para los médicos de CCA, por el seguimiento debe darse en centros especializados en CCA al menos 1 vez al año e incluir ecocardiografía, ECG, análisis de sangre y pruebas de ejercicio. Los intervalos para la RMC y la ecografía hepática (o TC) deben decidirse según cada caso individual. En los adultos es razonable realizar una evaluación hepática basal con RM en la primera visita, para guiar la frecuencia y el modo del seguimiento de acuerdo con el grado de los cambios hepáticos preexistentes. Además, se debe considerar una evaluación hepática anual que incluya ecografía hepática y determinación de alfa-fetoproteína tras consultarlo con los servicios locales de hepatología. La evaluación completa es obligatoria en pacientes con manifestaciones del complejo de fracaso de Fontan, y se debe prestar especial atención para excluir incluso las obstrucciones menores al flujo cavopulmonar y el retorno venoso pulmonar, ya que pueden tener un impacto hemodinámico importante.
4.15.7Consideraciones adicionales- •
Ejercicio/deporte: tras la operación de Fontan, los pacientes tienen una limitación importante a su capacidad de ejercicio como parte de su circulación. Según las recomendaciones recientes, se debe aconsejar la práctica de ejercicio aeróbico limitada por los síntomas para mejorar la fuerza muscular y la calidad de vida24.
- •
Embarazo: está contraindicado para las pacientes con circulación de Fontan y alguna complicación. El embarazo exitoso es posible para pacientes seleccionadas, aunque se asocia con una morbilidad materna importante, sobre todo insuficiencia cardiaca, arritmias y complicaciones tromboembólicas. Se debe considerar la administración de anticoagulantes, sopesado frente al riesgo hemorrágico, que es mayor en estas pacientes. Es obligatorio realizar una monitorización intensiva, periodo posparto incluido. Hay una tasa de aborto espontáneo de un 27–55% y altas tasas de prematuridad y restricción del crecimiento intrauterino. Se desconoce si la sobrecarga de volumen que conlleva el embarazo tiene un efecto adverso en el resultado a largo plazo de mujeres con un solo ventrículo.
- •
Profilaxis de la EI: se recomienda solo para pacientes con reintervención de Fontan reciente (< 6 meses), cianosis, válvula protésica, derrame del parche residual o endocarditis previa.
Las anomalías coronarias incluyen la coronaria anómala con origen en la arteria pulmonar (CAAP), el origen aórtico anómalo de una arteria coronaria (OAAAC) y las fístulas coronarias.
4.16.1.1Arteria coronaria anómala con origen en la arteria pulmonarAunque muchas anomalías coronarias congénitas son benignas, los estudios sobre la historia natural de las CAAP han documentado un resultado pobre en ausencia de tratamiento318. La CAAP resulta en una concentración baja de oxígeno en la arteria coronaria, síndrome de robo coronario e infarto de miocardio. La ALCAPA se puede presentar en forma de infarto de miocardio silente o sintomático, disfunción del VI, TV o incluso MSC. Los pacientes también pueden sufrir sobrecarga de volumen por cortocircuito I-D que causa síntomas de insuficiencia cardiaca. Por el contrario, la ARCAPA se suele diagnosticar incidentalmente. Se prefiere la reparación del sistema coronario dual, incluida la transferencia del botón coronario con o sin un injerto de interposición. La cirugía de revascularización coronaria (CABG) con cierre de la CAAP debe reservarse para casos en los que la transferencia coronaria no es factible.
4.16.1.2Origen aórtico anómalo de una arteria coronariaNo hay estudios sobre la historia natural de la enfermedad en pacientes con OAAAC no tratados. El tratamiento aún es tema de debate, sobre todo para pacientes con un trayecto interarterial de una arteria coronaria anómala. La evaluación del riesgo de MSC es difícil por la ausencia de datos. Las series de autopsias muestran que la mayoría de los pacientes son jóvenes (< 35 años) y mueren durante o inmediatamente después de la práctica de ejercicio. Se ha demostrado fibrosis miocárdica, lo que indica que la isquemia puede tomar parte. La arteria coronaria izquierda que surge del seno opuesto (derecho) es menos común, pero más maligna que la arteria coronaria derecha del seno izquierdo. El orificio alto, la estenosis ostial, el orificio en forma de hendidura/boca de pez, el despegue en ángulo agudo, el trayecto intramural y su longitud, o el trayecto interarterial y la hipoplasia de la arteria coronaria proximal se han asociado con isquemia miocárdica y se han propuesto como factores de riesgo319–322.
La estratificación del riesgo debe incluir también la edad (< 35 años) y el grado de ejercicio (p. ej., deportes de competición). Hay muy poca evidencia que indique que la cirugía en pacientes de mediana edad asintomáticos mejore la supervivencia o modifique el riesgo de MSC323,324.
4.16.1.3Fístulas coronariasUna fístula coronaria, ya sea congénita o adquirida, es una conexión anormal entre una arteria coronaria y la cámara cardiaca o un vaso. Las fístulas pequeñas tienen un pronóstico bueno sin tratamiento. Las fístulas medianas o grandes se asocian con complicaciones a largo plazo (angina, infarto de miocardio, arritmias, insuficiencia cardiaca y endocarditis). La presencia de síntomas, complicaciones y un cortocircuito relevante son las indicaciones principales para el cierre percutáneo o quirúrgico.
4.16.1.4Evaluación diagnósticaLa TCC es la técnica de elección para la evaluación de la anatomía de alto riesgo, que incluye características como un trayecto intramural y anomalías en el orificio (orificio en forma de hendidura, despegue en ángulo agudo, orificio > 1cm por encima de la unión sinotubular).
La evaluación de la isquemia inducida por estrés mediante técnicas de imagen avanzadas es la clave para la toma de decisiones.
Recomendaciones para el tratamiento de pacientes con arterias coronarias anómalas
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Se recomienda la obtención de imágenes funcionales no farmacológicas (p. ej., estudio nuclear, ecocardiografía o RMC con estrés físico) de los pacientes con anomalías coronarias para confirmar/excluir isquemia miocárdica | I | C |
| Coronaria anómala con origen en la arteria pulmonar | ||
| Se recomienda la cirugía para los pacientes con ALCAPA | I | C |
| Se recomienda la cirugía para los pacientes con ARCAPA y síntomas atribuibles a la coronaria anómala | I | C |
| Se debe considerar la cirugía para los pacientes asintomáticos con ARCAPA y disfunción ventricular o isquemia miocárdica atribuible a la anomalía coronaria | IIa | C |
| Origen aórtico anómalo de una arteria coronaria | ||
| Se recomienda la cirugía para los pacientes con OAAAC y síntomas típicos de angina que se presentan con evidencia de isquemia miocárdica inducida por estrés en un territorio equivalente o anatomía de alto riesgoc | I | C |
| Para los pacientes con OAAAC (derecho o izquierdo) asintomáticos con evidencia de isquemia miocárdica, se debe considerar la cirugía | IIa | C |
| Para los pacientes con OAAACI asintomáticos sin evidencia de isquemia miocárdica pero con anatomía de alto riesgoc, se debe considerar la cirugía | IIa | C |
| Para los pacientes con OAAAC sintomáticos, se podría considerar la cirugía incluso cuando no haya evidencia de isquemia miocárdica ni anatomía de alto riesgoc | IIb | C |
| Para los pacientes con OAAACI asintomáticos sin isquemia miocárdica ni anatomía de alto riesgoc, se podría considerar la cirugía cuando se presentan a una edad joven (< 35 años) | IIb | C |
| La cirugía no está indicada para los pacientes con OAAACD asintomáticos sin isquemia miocárdica ni anatomía de alto riesgoc | III | C |
ALCAPA: coronaria izquierda anómala desde la arteria pulmonar; ARCAPA: coronaria derecha anómala desde la arteria pulmonar; OAAAC: origen aórtico anómalo de una arteria coronaria; OAAACD: origen aórtico anómalo de la coronaria derecha; OAAACI: origen aórtico anómalo de la coronaria izquierda; RMC: resonancia magnética cardiovascular.
aClase de recomendación.
bNivel de evidencia.
cLa anatomía de alto riesgo incluye características tales como un trayecto intramural y anomalías del orificio (orificio en forma de hendidura, despegue en ángulo agudo, orificio > 1cm por encima de la unión sinotubular).
Las indicaciones para la cirugía se resumen en la tabla «Recomendaciones para el tratamiento de pacientes con anomalías coronarias».
5INDICADORES DE CALIDADEl objetivo general de esta nueva edición de la guía para el tratamiento de las CCA es ayudar a los cuidadores en su práctica diaria en beneficio de los pacientes. Es importante analizar la adaptación y el cumplimiento de esta guía en la práctica clínica, un proceso que puede cuantificarse con indicadores de calidad (QI).
Los QI son conjuntos de medidas que permiten la cuantificación del cumplimiento de las recomendaciones de la guía y proporcionan un mecanismo para medir las oportunidades de mejorar la atención y los resultados cardiovasculares325. Los QI muestran importantes diferencias con las guías de práctica clínica. Mientras que las guías ofrecen recomendaciones para que la atención se aplique de manera prospectiva a pacientes individuales, los QI se aplican retrospectivamente a un grupo de pacientes para evaluar si se les brindó o no un determinado proceso de atención325.
Los QI derivan de la evidencia, son factibles, interpretables de un modo concreto y prácticos326. Su objetivo es mejorar la calidad de la atención médica y los emplean cada vez más las autoridades sanitarias, las organizaciones profesionales, los contribuyentes y la población general327–329.
El proceso de desarrollo y definición de QI para el abordaje de aspectos concretos de las CCA se inició durante el proceso de redacción de la guía y los resultados se publicarán más adelante en un documento específico.
6LAGUNAS EN LA EVIDENCIA6.1Aspectos generales6.1.1Planificación del cuidado y evaluación del paciente- •
Los defectos cardiacos congénitos se han clasificado arbitrariamente en lesiones de diferente complejidad (leves, moderadas y graves) (tabla 4). La validez de esta clasificación para el tratamiento clínico y la estratificación del riesgo se debe investigar en grandes registros.
- •
Es necesario definir el volumen mínimo de pacientes atendidos en cada centro de expertos de CCA y el personal necesario para tener unos resultados óptimos.
- •
Es necesario establecer medidas de resultados relevantes, además de la mortalidad, para cuantificar la calidad de la atención.
- •
Debe definirse mejor el papel de las neurohormonas para estimar la gravedad de la enfermedad y el momento de las intervenciones.
- •
La fisiopatología de la insuficiencia cardiaca, sobre todo en pacientes con VD sistémico y circulación de Fontan, no se conoce bien y es necesario llevar a cabo estudios para mejorar su prevención y tratamiento.
- •
Deben definirse mejor las indicaciones para la aplicación del tratamiento estándar de la insuficiencia cardiaca, tanto en el contexto agudo como en el crónico.
- •
Se debe estudiar mejor aspectos relacionados con la predicción y el curso de la insuficiencia cardiaca para mejorar las indicaciones y el momento del DAI/trasplante.
- •
Se necesitan registros prospectivos de colaboración internacional a gran escala sobre tratamiento médico y con dispositivos en las CCA y se requiere apoyo para su implementación.
- •
Se necesitan sistemas de puntuación específicos para evaluar la indicación de anticoagulación en el contexto de las arritmias auriculares para las CC moderada y compleja.
- •
La ablación con catéter dirigida a los istmos anatómicos de conducción lenta ha sido muy eficaz para controlar la TV monomórfica en la TdF. Hacen falta más estudios para establecer la utilidad del mapeo con catéter en la estratificación del riesgo individualizada de pacientes sin TV espontánea en la TdF y los defectos relacionados.
- •
Preocupa la posible pérdida de acceso a los istmos anatómicos de conducción lenta para la ablación con catéter después de la reparación de la TdF y otros defectos relacionados. Se debe estudiar si los pacientes sin TV documentada se benefician de la ablación preventiva antes o durante la reparación de la TdF.
- •
Las indicaciones de estimulación y TRC para pacientes con CCA se derivan principalmente de adultos con corazones anatómicamente normales y miocardiopatía isquémica o dilatada y no están adaptadas a la diversidad de sustratos estructurales y funcionales de las CC. Es necesario seguir investigando cuáles son los candidatos óptimos para la TRC y los sitios de estimulación adecuados en las diferentes CC.
- •
Es necesario hacer más estudios sobre el impacto del tratamiento médico de la HAP en la supervivencia de pacientes con síndrome de Eisenmenger.
- •
El papel del tratamiento combinado inicial en la HAP-CC requiere más atención.
- •
Hay poca experiencia sobre el tratamiento con prostaciclina en pacientes con HAP-CC y sería necesario realizar más estudios.
- •
El beneficio de la anticoagulación sistemática en ausencia de factores importantes de riesgo de complicaciones tromboembólicas (p. ej., arritmias auriculares) es controvertido y requiere más estudios.
- •
El resultado a largo plazo, especialmente las arritmias, tras el cierre del dispositivo requiere más estudios.
- •
El impacto del cierre de la derivación en el resultado a largo plazo de los pacientes con HAP sigue siendo incierto; se requiere más investigación para definir los umbrales para las recomendaciones terapéuticas.
- •
Se debe definir mejor el momento óptimo para la intervención de pacientes asintomáticos con OTSVI grave.
- •
El implante percutáneo de la válvula aórtica evoluciona rápidamente; es necesario refinar su papel en las CCA.
- •
Según la guía de la ESC de 2018 para el tratamiento de la hipertensión arterial190, las definiciones de hipertensión en pacientes con CoA reparada son las mismas que en la hipertensión arterial general, y se debe tratar a los pacientes de acuerdo con las guías generales; falta evidencia para esta estrategia y se requieren estudios prospectivos.
- •
La estimación del riesgo de disección aórtica y la definición del umbral para la cirugía profiláctica en la EHAT basada solo en el diámetro es subóptima y requiere un enfoque más personalizado. Es necesario definir si el tipo de defecto genético subyacente es útil en esta estratificación.
- •
Los regímenes de tratamiento profiláctico actuales para pacientes con EHAT incluyen los bloqueadores beta y los ARA–II. Ninguno de ellos (ya sea en monoterapia o combinados) previene el crecimiento de la aorta. Es necesario seguir buscando nuevas dianas terapéuticas.
- •
Se deben refinar los criterios para la reparación concomitante de la VT en el momento de la cirugía del TSVD.
- •
Es necesario mejorar la identificación de los pacientes con OTSVD de bajo gradiente que tienen estenosis grave y pueden mejorar mediante intervención.
- •
Es preciso investigar los criterios para identificar a los pacientes con OTSVD que puedan beneficiarse de una intervención para la IP residual.
- •
El papel del estudio EF para la estratificación del riesgo de MSC es controvertido en pacientes con TdF y se debe estudiar.
- •
Es necesario mejorar la identificación de los pacientes con Ebstein asintomáticos e IT grave que puedan beneficiarse de la cirugía de la VT.
- •
Es preciso mejorar la identificación de pacientes con Ebstein y riesgo de arritmias tardías que pueden poner en peligro la vida.
- •
Es necesario definir el momento óptimo para la SVP en pacientes asintomáticos con IP relevante.
- •
Se requieren estudios de seguimiento a largo plazo después del IPVP para aumentar el conocimiento sobre la durabilidad de la válvula, las consecuencias de las fracturas del stent y la aparición de endocarditis.
- •
Debe mejorarse la identificación de pacientes con TdF y riesgo de arritmias tardías con riesgo vital que se beneficiarían del implante de un DAI como estrategia de prevención primaria.
- •
Se debe establecer el efecto del tratamiento médico en la dilatación o disfunción del VD en pacientes con TdF.
- •
Es preciso estudiar mejor el beneficio potencial del tratamiento médico clásico para la insuficiencia cardiaca y la estimulación biventricular en pacientes con un VD sistémico después de un procedimiento de switch auricular.
- •
Es necesario refinar la estratificación del riesgo de MSC y las indicaciones para el implante de DAI tras un switch arterial.
- •
Tras un procedimiento de switch arterial, se debe definir mejor el riesgo de disección/rotura de los aneurismas de la raíz neoaórtica para refinar las recomendaciones de la cirugía profiláctica.
- •
Se requiere un seguimiento a largo plazo después del switch arterial para estudiar el riesgo de EC después del reimplante de las arterias coronarias en la raíz neoaórtica.
- •
Se necesita disponer de más datos para definir el momento óptimo de la sustitución de la VT en pacientes asintomáticos con IT grave.
- •
Se debe investigar el beneficio potencial de colocar una banda pulmonar para conservar la función ventricular sistémica.
- •
Es necesario investigar el papel de la medicación, incluidos los vasodilatadores pulmonares, en pacientes con circulación de Fontan.
- •
Se deben establecer los efectos del embarazo en los resultados maternos a largo plazo.
- •
Es preciso investigar los determinantes fisiológicos (incluido el papel del sistema linfático) en los resultados a largo plazo en pacientes con circulación de Fontan.
- •
Se requieren más estudios pata identificar a los pacientes adultos con anomalías coronarias (OAAAC, CAAP) que están en riesgo de MSC y podrían obtener beneficios de la cirugía en la edad adulta.
- •
Satisfacer las necesidades de los pacientes con CCA tiene requisitos especiales tanto estructurales como organizativos de la asistencia sanitaria.
- •
Las técnicas de imagen multimodales son clave para la evaluación adecuada de la anatomía general y la función ventricular y valvular, así como para cuantificar el flujo sanguíneo y la distribución de la perfusión.
- •
La prueba de ejercicio es una medio importante para determinar el momento de las intervenciones y reintervenciones.
- •
El cateterismo cardiaco sigue siendo clave para la evaluación hemodinámica, en particular, de la PAP y la resistencia vascular.
- •
El tratamiento clave para la insuficiencia cardiaca de pacientes con CCA sigue siendo prevenirla optimizando la hemodinámica y el ritmo cardiaco, lo que requiere un seguimiento sistemático en centros especializados que faciliten la intervención cuando sea oportuno.
- •
En una circulación biventricular, se puede extrapolar el tratamiento estándar de la insuficiencia cardiaca a los pacientes con CCA y VI sistémico; la extrapolación a los pacientes con VD sistémico también es posible, aunque en estos casos el beneficio sigue siendo incierto. La fisiopatología de los pacientes con switch auricular, sobre todo cuando hay CUV y Fontan paliativo, difiere notablemente de una «circulación regular», por lo que el tratamiento estándar para la insuficiencia cardiaca debe aplicarse con precaución.
- •
Se recomienda la derivación a un centro con experiencia en CCA y trasplantes y la consulta con especialistas en CCA e insuficiencia cardiaca, especialmente en casos de CC de complejidad moderada y grave.
- •
Se debe examinar una posible causa reversible de arritmia y las anomalías hemodinámicas nuevas o residuales en todos los pacientes.
- •
El objetivo es mantener el ritmo sinusal en la mayoría de los pacientes con CCA.
- •
El tratamiento óptimo de las arritmias crónicas exige la derivación a un centro especializado en CCA que cuente con un equipo multidisciplinario.
- •
Los casos clínicos de arritmias documentadas o con riesgo alto de arritmias tras el procedimiento considerados para (re)intervenciones percutáneas o quirúrgicas deben discutirse en un contexto multidisciplinario con experiencia en intervenciones y tratamiento invasivo de arritmias.
- •
La HAP en las CCA es una enfermedad progresiva con mal pronóstico.
- •
Se recomienda sospechar HAP después del cierre del defecto y evaluar periódicamente la HAP en pacientes con cortocircuitos.
- •
Se requiere un tratamiento proactivo de todos los pacientes con HAP, incluidos aquellos con síndrome de Eisenmenger.
- •
Se debe desaconsejar el embarazo a las mujeres con CCA y HAP confirmada.
- •
Los pacientes cianóticos presentan un trastorno multisistémico y corren el riesgo de sufrir complicaciones hemorrágicas y trombóticas, lo que produce un dilema terapéutico.
- •
Deben evitarse las flebotomías por sistema, ya que ponen a los pacientes en riesgo de anemia ferropénica y complicaciones cerebrovasculares. La flebotomía terapéutica solo está indicada en presencia de síntomas de hiperviscosidad moderados/graves.
- •
Los pacientes cianóticos tienen una fisiopatología muy equilibrada pero frágil, y cualquier intervención los pone en un riesgo alto. Por lo tanto, todas las intervenciones deben realizarse en un centro especializado en CCA.
- •
Las medidas profilácticas son el pilar de la atención para prevenir y evitar complicaciones.
- •
Las decisiones terapéuticas requieren una evaluación cuidadosa de la sobrecarga de volumen ventricular y la circulación pulmonar.
- •
En pacientes con signos no invasivos de PAP elevada, es obligatorio un cateterismo cardiaco con evaluación de la RVP.
- •
En presencia de RVP ≥ 5 UW, se debe evitar el cierre de la CIA. Solo se puede considerar el cierre del DSV y DAP en pacientes seleccionados con cortocircuito relevante tras la evaluación cuidadosa en un centro especializado en CCA e HP.
- •
El cierre con dispositivo es el tratamiento de elección cuando sea técnicamente factible.
- •
Las principales indicaciones para cirugía siguen siendo los síntomas y la disfunción del VI.
- •
Los pacientes con obstrucción grave que no refieren síntomas deben hacer una prueba de esfuerzo para confirmar el estado asintomático.
- •
En la EA valvular congénita, debe excluirse la enfermedad aórtica asociada (dilatación de la aorta ascendente o CoA).
- •
Es fundamental la medición correcta de la presión arterial (brazo derecho, ambulatorio) en el seguimiento de los pacientes con CoA.
- •
La decisión de (re)intervenir depende de la presión arterial, el gradiente y la morfología de la estenosis.
- •
La colocación de stents es el tratamiento de elección cuando sea técnicamente factible.
- •
Es esencial supervisar de por vida a todos los pacientes con EHAT mediante imágenes de toda la aorta, así como la evaluación de la función valvular y miocárdica.
- •
El diámetro aórtico para la indicación de la cirugía depende de la enfermedad subyacente y la presencia de factores de riesgo.
- •
La OTSVO puede estar sobrestimada por la velocidad del flujo a través de la obstrucción, particularmente cuando el estrechamiento es alargado o hay estenosis en serie (p. ej., subvalvular y valvular). Por consiguiente, se requiere una verificación cruzada con la presión del VD calculada a partir de la velocidad de la IT.
- •
El cateterismo intervencionista es el tratamiento de elección para pacientes con EP valvular no displásica (valvuloplastia con balón) o EP periférica (a menudo con implante de stent).
- •
La indicación de intervención es más restrictiva cuando se requiere un reemplazo valvular debido a sus implicaciones a largo plazo (complicaciones y necesidad de reintervención).
- •
El momento óptimo para la cirugía sigue siendo un reto; esta operación solo deben realizarla cirujanos con experiencia específica en esta lesión.
- •
La reparación valvular es la técnica preferida siempre que sea factible.
- •
La IP o la OTSVD relevantes, la disfunción del VD y el VI y las arritmias son complicaciones frecuentes a largo plazo.
- •
Los posibles factores de riesgo de cualquier arritmia ventricular y MSC en la TdF son: duración del QRS > 180ms, disfunción sistólica o diastólica del VI, disfunción del VD, TV inducible en la estimulación eléctrica programada y el antecedente de arritmia auricular.
- •
El momento óptimo para la intervención en la IP grave asintomática sigue siendo un reto. La normalización del tamaño del VD se vuelve poco probable cuando el índice de volumen telediastólico excede los 160 ml/m2, pero este punto de corte para la reintervención puede no correlacionarse con el beneficio clínico.
- •
El IPVP se ha convertido en el tratamiento de elección para la reintervención del TSVD cuando sea anatómicamente factible.
- •
La insuficiencia ventricular sistémica, la insuficiencia secundaria de la válvula AV sistémica, las arritmias y la estenosis del bafle con o sin derrame son complicaciones frecuentes a largo plazo que se debe tratar después de la operación de switch auricular.
- •
El resultado de la morbilidad ha mejorado notablemente con la introducción de la operación de switch arterial. La dilatación de la raíz neoaórtica con o sin insuficiencia significativa de la válvula neoaórtica, EP supravalvular y estenosis de la rama pulmonar ocurren principalmente durante la infancia, pero pueden necesitar reintervención en la edad adulta.
- •
La aparición de disfunción sistólica del VI de novo o arritmias después del switch arterial requieren una evaluación completa que excluya estenosis ostiales/proximales de las arterias coronarias reimplantadas.
- •
El fracaso del conducto VD-AP (estenosis, regurgitación o ambas) es la complicación predominante a largo plazo, y requiere reintervención tras la operación de tipo Rastelli.
- •
La insuficiencia del VD sistémico, la insuficiencia de la válvula AV sistémica, el bloqueo AV y la arritmia auricular son complicaciones tardías frecuentes.
- •
La insuficiencia de la válvula AV sistémica es un determinante importante del resultado tardío y, cuando es grave, debe abordarse antes de que la función sistémica del VD se vea afectada.
- •
Aunque la calidad de vida de muchos pacientes con Fontan es buena, todos requieren un intenso seguimiento regular en un centro especializado en CCA, ya que corren el riesgo de sufrir múltiples complicaciones graves, como arritmia, insuficiencia cardiaca, enfermedad hepática y enteropatía pierdeproteínas.
- •
Es imprescindible mantener una PAP baja para el buen funcionamiento de la circulación de Fontan; se recomienda un umbral bajo para la evaluación hemodinámica invasiva cuando se sospeche disfunción o se produzcan complicaciones.
- •
Las arritmias se toleran mal y requieren una acción inmediata.
- •
Es factible el embarazo de pacientes seleccionadas con circulación de Fontan en buen funcionamiento, pero con un alto riesgo de aborto espontáneo, y se debe atender la gestación en un centro especializado en CCA.
- •
Es obligatorio vigilar los problemas hepáticos de todos los pacientes con Fontan.
- •
La TCC es la técnica preferida para la evaluación de la anatomía de alto riesgo, incluidas características como un trayecto intramural y anomalías en el orificio (orificio en forma de hendidura, despegue en ángulo agudo u orificio > 1cm por encima de la unión senotubular).
- •
La valoración de la isquemia inducida por estrés mediante técnicas de imagen avanzadas con estrés físico es la clave para la toma de decisiones.
- •
En pacientes con fístula coronaria, los síntomas, las complicaciones y los cortocircuitos relevantes son las principales indicaciones para el cierre percutáneo o quirúrgico.
| Recomendaciones | Clasea | Nivelb |
|---|---|---|
| Tratamiento de las arritmias en las cardiopatías congénitas del adulto | ||
| Los pacientes con CC de complejidad moderada y grave (tabla 4) y arritmias documentadas deben ser derivados a un centro que cuente con un equipo multidisciplinario y tenga experiencia en el tratamiento de pacientes con CCA y las arritmias relacionadas | I | C |
| Los pacientes con CC y arritmias documentadas o con riesgo alto de arritmias tras el procedimiento (p. ej., cierre de CIA a una edad más avanzada) para quienes se consideran (re)intervenciones percutáneas o quirúrgicas deben ser derivados a un centro que cuente con un equipo multidisciplinario y que tenga experiencia en estas intervenciones y en el tratamiento invasivo de las arritmias | I | C |
| En la CC leve, se recomienda la ablación con catéter en lugar del tratamiento médico de larga duración para la TSV recurrente sostenida y sintomática (TRNAV, TRAV, TA y TRIA) o si la TSV puede estar relacionada con MSC (tabla 7) | I | C |
| La ablación con catéter está indicada como tratamiento complementario a los DAI para los pacientes con TV monomórfica recurrente, TV incesante o tormenta eléctrica no tratale con fármacos o reprogramación del DAI | I | C |
| Tras evaluar la causa del episodio y excluir las causas reversibles, está indicado el implante de DAI en adultos con CC que han sobrevivido a parada cardiaca por FV o TV no tolerada hemodinámicamente | I | C |
| Para los adultos con CC y TV sostenida, está indicado el implante de DAI después de evaluación hemodinámica y reparación cuando esté indicada. El estudio EF es necesario para identificar a los pacientes para quienes la ablación percutánea o quirúrgica pueda ser beneficiosa como tratamiento complementario o una posible alternativa razonable | I | C |
| Tratamiento de la hipertensión arterial pulmonar asociada con la cardiopatía congénita | ||
| Se recomienda desaconsejar el embarazo a las pacientes con CC e HP precapilar confirmada | I | C |
| Se recomienda la evaluación del riesgo de todos los pacientes con HAP-CC | I | C |
| Para los pacientes con riesgo bajo e intermedio, lesiones simples reparadas y HP precapilar, se recomienda el tratamiento oral combinado inicial o secuencial; para los pacientes con alto riesgo el tratamiento debe ser uno combinado inicial que incluya prostanoides parenterales | I | A |
| Comunicación interauricular (congénita y residual) | ||
| Los pacientes con evidencia de sobrecarga de volumen del VD y sin HAP (sin signos no invasivos de elevación de PAP o confirmación invasiva de RVP < 3 UW en caso de que los haya) o enfermedad del VI deben someterse a cierre de la CIA con independencia de los síntomas | I | B |
| El cierre con dispositivo es el método de elección para el cierre de la CIA de tipo ostium secundum siempre que sea posible | I | C |
| Para los pacientes de edad avanzada no aptos para el cierre con dispositivo, se recomienda sopesar cuidadosamente el riesgo quirúrgico frente al beneficio potencial del cierre de la CIA | I | C |
| Es imprescindible la medición invasiva de la RVP en pacientes que presenten signos no invasivos de elevación de la PAP | I | C |
| En pacientes con enfermedad del VI, se recomienda realizar una prueba con balón y sopesar cuidadosamente el beneficio de eliminar el cortocircuito I-D frente al posible impacto negativo del cierre debido a un aumento en la presión de llenado (considerando cierre, cierre fenestrado y no cerrar) | I | C |
| Se debe evitar el cierre de la CIA de pacientes con fisiología de Eisenmenger o con HAP y RVP ≥ 5 WU a pesar del tratamiento dirigido a la HAP o cuando se produzca desaturación durante el ejercicio | III | C |
| Defecto sel septo ventricular (congénito y residual) | ||
| Los pacientes con sobrecarga de volumen VI que no presentan HAP (ausencia de signos no invasivos de elevación de PAP o confirmación invasiva de RVP < 3 UW en caso de tales signos) deben someterse a cierre del DSV con independencia de los síntomas | I | C |
| Se debe evitar el cierre del DSV de los pacientes con fisiología de Eisenmenger o con HAP grave (RVP ≥ 5 WU) que sufran desaturación durante el ejercicio | III | C |
| Defecto del septo auriculoventricular | ||
| Se debe evitar la reparación quirúrgica de pacientes con fisiología de Eisenmenger o con HAP grave (RVP ≥ 5 WU) que sufran desaturación durante el ejercicio | III | C |
| Se recomienda el cierre quirúrgico por un cirujano cardiaco experto en CC en caso de sobrecarga relevante de volumen del VD | I | C |
| Para los pacientes sintomáticos con insuficiencia de la válvula AV de moderada a grave, se recomienda la cirugía valvular, preferiblemente reparación de la válvula AV, por cirujanos cardiacos expertos en CC | I | C |
| Para los pacientes asintomáticos con insuficiencia grave de la válvula del lado izquierdo, se recomienda la cirugía de la válvula si la DTSVI es ≥ 45mm o la FEVI ≤ 60% tras haber excluido otras causas de disfunción del VI | I | C |
| Ductusarterioso persistente | ||
| Para los pacientes con sobrecarga del volumen del VI sin HAP (ausencia de signos no invasivos de elevación de PAP o confirmación invasiva de RVP < 3 UW en caso de que los haya), se recomienda cerrar el DAP con independencia de los síntomas | I | C |
| Se recomienda el cierre con dispositivo como el método de elección cuando sea técnicamente factible | I | C |
| El cierre del DAP debe evitarse en pacientes con fisiología de Eisenmenger o que sufran desaturación en las extremidades inferiores durante el ejercicio | III | C |
| Estenosis valvular aórtica | ||
| Para los pacientes sintomáticos con EA grave de alto gradiente (gradiente medio ≥ 40 mmHg), se recomienda la intervención | I | B |
| La intervención está indicada para los pacientes sintomáticos con EA grave de bajo flujo y bajo gradiente (gradiente medio < 40 mmHg) que presenten FE reducida y reserva contráctil que excluya EA seudograve | I | C |
| Para los pacientes asintomáticos con EA grave, está indicada la cirugía cuando sufran síntomas durante la prueba de ejercicio | I | C |
| La intervención está indicada para los pacientes asintomáticos con EA grave cuando haya disfunción sistólica del VI (FEVI < 50%), a menos que se deba a otras causas | I | C |
| Se recomienda la cirugía cuando los pacientes con EA grave se sometan a cirugía de la aorta ascendente u otra válvula o a CABG | I | C |
| Estenosis aórtica supravalvular | ||
| Para los pacientes con síntomas, espontáneos o en la prueba de ejercicio, y gradiente Doppler medio ≥ 40mmHg, se recomienda la cirugía | I | C |
| Para los pacientes con gradiente Doppler medio < 40mmHg, la cirugía está recomendada con los siguientes hallazgos:• Síntomas atribuibles a una obstrucción (disnea de esfuerzo, angina, síncope) o• Disfunción sistólica del VI (FE < 50%) sin otra explicación• Necesidad de cirugía por EC o valvulopatía relevantes | I | C |
| Estenosis subaórtica | ||
| Para los pacientes con síntomas, espontáneos o en la prueba de ejercicio, con un gradiente Doppler medio ≥ 40mmHg o IAo grave, se recomienda la cirugía | I | C |
| Coartación y recoartación aórtica | ||
| La reparación de la coartación o recoartación aórtica están indicadas para los pacientes hipertensos con aumento de la presión no invasiva entre las extremidades superiores e inferiores confirmada por una determinación invasiva (pico-pico ≥ 20 mmHg), preferentemente porpercutáneo (implante de stent) cuando sea técnicamente factible | I | C |
| Cirugía aórtica en las aortopatías | ||
| Está indicada la reparación de la válvula aórtica mediante técnicas de reimplante o remodelado por anuloplastia aórtica para los pacientes jóvenes con síndrome de Marfan o EHAT relacionada con dilatación de la raíz aórtica y válvula aórtica tricúspide siempre que la realicen cirujanos experimentados | I | C |
| Está indicada la cirugía para los pacientes con síndrome de Marfan y enfermedad de la raíz aórtica con un diámetro máximo del seno aórtico ≥ 50 mm | I | C |
| Obstrucción del tracto de salida del ventrículo derecho | ||
| La valvuloplastia con balón es la intervención de elección para la EP si es anatómicamente factible | I | C |
| Si no se requiere sustitución valvular, se recomienda la intervención de la OTSVD a cualquier nivel con independencia de los síntomas cuando hayaestenosis grave (gradiente Doppler máximo > 64 mmHg) | I | C |
| Cuando la sustitución quirúrgica de la válvula sea la única opción, la cirugía está indicada para los pacientes con estenosis grave que están sintomáticos | I | C |
| Cuando la sustitución quirúrgica de la válvula sea la única opción, la cirugía está indicada para los pacientes con estenosis grave que están asintomáticos en presencia de:• Reducción objetiva de la capacidad de ejercicio o• Función del VD decreciente o progresión de la IT a ≥ moderada• Presión sistólica del VD > 80 mmHg• Cortocircuito D-I vía CIA o DSV | I | C |
| Anomalía de Ebstein | ||
| Se recomienda la reparación quirúrgica para los pacientes sintomáticos con IT grave o capacidad de ejercicio deteriorada medida por PECP | I | C |
| Se recomienda que realice la reparación quirúrgica un cirujano especialista en CC con experiencia específica en cirugía de Ebstein | I | C |
| Si también hay una indicación para cirugía de la VT, se recomienda el cierre quirúrgico de la CIA/FOP en el momento de reparar la válvula si se tolera hemodinámicamente | I | C |
| Para los pacientes con arritmias relevantes o preexcitación en el ECG, se recomienda una prueba electrofisiológica, seguida de terapia con ablación de ser posible o tratamiento quirúrgico de las arritmias en caso de cirugía cardiaca programada | I | C |
| Tras reparación de la tetralogía de Fallot | ||
| Se recomienda la SVP para los pacientes sintomáticos con IP grave o OTSVD al menos moderada | I | C |
| Se prefiere el cateterismo intervencionista (IPVP) si es anatómicamente factible para los pacientes que no tienen tracto de salida nativo | I | C |
| Transposición de las grandes arterias tras la operación deswitchauricular | ||
| Para los pacientes sintomáticos con obstrucción venosa pulmonar, se recomienda la reparación quirúrgica (el cateterismo intervencionista casi nunca es posible) | I | C |
| Para los pacientes sintomáticos con estenosis del bafle no susceptibles de tratamiento percutáneo, se recomienda la reparación quirúrgica | I | C |
| Para los pacientes sintomáticos con derrames del bafle no susceptibles de tratamiento con stent, se recomienda la cirugía | I | C |
| No se recomienda la banda en la AP para los pacientes adultos para entrenamiento del VI con posterior switch arterial | III | C |
| Para los pacientes sintomáticos con estenosis del bafle, se recomienda el implante de stent | I | C |
| Para los pacientes sintomáticos con derrames del bafle y cianosis considerable en reposo o ejercicio y aquellos con firme sospecha de embolia paradójica, se recomienda el implante de stent recubierto o el cierre con dispositivo | I | C |
| Para los pacientes con derrame del bafle y síntomas secundarios al cortocircuito I-D, se recomienda el implante de stent recubierto o el cierre con dispositivo | I | C |
| Transposición de las grandes arterias tras la operación deswitcharterial | ||
| Se recomienda el implante de stent o cirugía, dependiendo del sustrato, para la estenosis de la arteria coronaria que causa isquemia | I | C |
| Transposición de las grandes arterias congénitamente corregida | ||
| Para los pacientes sintomáticos con IT grave y función del VD sistémico conservada o levemente deteriorada (FE > 40%), se recomienda la sustitución de la VT | I | C |
| Conductos del ventrículo derecho a la arteria pulmonar | ||
| Los pacientes sintomáticos con presión sistólica del VD > 60mmHg (puede ser inferior en caso de flujo reducido) o RP grave deben someterse a intervención, preferiblemente percutánea (IPVP) si es anatómicamente factible | I | C |
| Corazón univentricular | ||
| Para los adultos con CUV no operado o paliado, se recomienda una evaluación cuidadosa en centros especializados que incluya técnicas multimodales de imagen, así como una evaluación diagnóstica invasiva para decidir si pueden beneficiarse de un procedimiento quirúrgico o intervencionista | I | C |
| Tras operación de Fontan | ||
| La arritmia auricular sostenida con conducción AV rápida es una urgencia médica y debe tratarse de inmediato con cardioversión eléctrica | I | C |
| La anticoagulación está indicada en presencia de (o con antecedentes de) trombo auricular, arritmias auriculares o episodios tromboembólicos | I | C |
| Se recomienda asesorar contra el embarazo a las mujeres con circulación de Fontan y cualquier complicación | I | C |
| Se recomienda el cateterismo cardiaco a un umbral bajo en casos de edema inexplicable, deterioro de la capacidad de ejercicio, arritmia de nueva aparición, cianosis y hemoptisis | I | C |
| Arterias coronarias anómalas | ||
| Se recomienda la obtención de imágenes funcionales no farmacológicas (p. ej., estudio nuclear, ecocardiografía o RMC con estrés físico) de los pacientes con anomalías coronarias para confirmar/excluir isquemia miocárdica | I | C |
| Coronaria anómala con origen en la arteria pulmonar | ||
| Se recomienda la cirugía en pacientes con ALCAPA. | I | C |
| Se recomienda la cirugía para los pacientes con ARCAPA y síntomas atribuibles a la coronaria anómala | I | C |
| Origen aórtico anómalo de una arteria coronaria | ||
| Se recomienda la cirugía para los pacientes con OAAAC, síntomas típicos de angina y evidencia de isquemia miocárdica inducida por estrés en un territorio equivalente o que tienen anatomía de alto riesgo | I | C |
| La cirugía no está indicada para los pacientes con OAAACD asintomáticos sin isquemia miocárdica ni anatomía de alto riesgo | III | C |
ALCAPA: coronaria izquierda anómala desde la arteria pulmonar; ARCAPA: coronaria derecha anómala desde la arteria pulmonar; AV: auriculoventricular; CABG: cirugía de revascularización coronaria; CC: cardiopatía congénita; CCA: cardiopatía congénita del adulto; CIA: comunicación interauricular; CUV: corazón univentricular; DAI: desfibrilador automático implantable; DAP: ductus arterioso persistente; D-I: derecho-izquierdo; DSAV: defecto del septo auriculoventricular; DSV: defecto del septo ventricular; DTSVI: diámetro telesistólico del VI; EA: estenosis aórtica; EC: enfermedad coronaria; ECG: electrocardiograma; EF: electrofisiológico; EHAT: enfermedad hereditaria de la aorta torácica; EP: estenosis pulmonar; FE: fracción de eyección; FEVI: fracción de eyección del VI; FOP: foramen oval permeable; FV: fibrilación ventricular; HAP: hipertensión arterial pulmonar; HAP-CC: hipertensión arterial pulmonar asociada con cardiopatía congénita; HP: hipertensión pulmonar; I-D: izquierda-derecha; IP: insuficiencia pulmonar; IPVP: implante percutáneo de la válvula pulmonar; IT: insuficiencia tricuspídea; MSC: muerte súbita cardiaca; OAAAC: origen aórtico anómalo de una arteria coronaria; OAAACD:origen aórtico anómalo de la coronaria derecha; OTSVD: obstrucción del tracto de salida del ventrículo derecho; PAP: presión arterial pulmonar; IAo: insuficiencia aórtica; RMC: resonancia magnética cardiovascular; RVP: resistencia vascular pulmonar; SVP: sustitución de válvula pulmonar; TA: taquicardia auricular; TRAV: taquicardia por reentrada auriculoventricular; TRNAV: taquicardia por reentrada del nódulo auriculoventricular; TRIA: taquicardia por reentrada intrauricular; TSV: taquicardia supraventricular; TV: taquicardia ventricular; VD: ventrículo derecho; VI: ventrículo izquierdo; VT: válvula tricúspide.
aClase de recomendación.
bNivel de evidencia.
Entidades de la ESC que han participado en la elaboración de este documento:
Asociaciones: Association for Acute Cardiovascular Care (ACVC), Association of Cardiovascular Nursing & Allied Professions (ACNAP), European Association of Cardiovascular Imaging (EACVI), European Association of Preventive Cardiology (EAPC), European Association of Percutaneous Cardiovascular Interventions (EAPCI), European Heart Rhythm Association (EHRA) y Heart Failure Association (HFA).
Consejos: Council for Cardiology Practice, Council of Cardio-Oncology y Council on Valvular Heart Disease.
Grupos de Trabajo: Adult Congenital Heart Disease, Aorta and Peripheral Vascular Diseases, Cardiovascular Pharmacotherapy, Cardiovascular Surgery, Development Anatomy and Pathology, e-Cardiology y Pulmonary Circulation and Right Ventricular Function.
El contenido de esta GPC de la ESC se ha publicado para uso personal y educativo solamente. No se autoriza su uso comercial. No se autoriza la traducción o reproducción de ningún fragmento de esta guía sin la autorización escrita de la ESC. La autorización se solicitará por escrito a Oxford University Press, editorial de European Heart Journal, o a los representantes autorizados de la ESC (journals.permissions@oup.com).
Descargo de responsabilidad. Esta guía recoge la opinión de la ESC y se ha elaborado tras el estudio minucioso de los datos y la evidencia disponibles hasta la fecha. La ESC no es responsable en caso de que exista alguna contradicción, discrepancia o ambigüedad entre la GPC de la ESC y cualquier otra recomendación oficial o GPC publicada por autoridades relevantes de la sanidad pública, particularmente en lo que se refiere al buen uso de la atención sanitaria y las estrategias terapéuticas. Se espera que los profesionales de la salud tengan en consideración esta GPC a la hora de tomar decisiones clínicas, así como al implementar estrategias médicas preventivas, diagnósticas o terapéuticas. No obstante, esta guía no anula la responsabilidad individual de cada profesional al tomar las decisiones oportunas relativas a cada paciente, de acuerdo con dicho paciente y, cuando fuera necesario, con su tutor o representante legal. Además, las GPC de la ESC no eximen al profesional médico de su obligación ética y profesional de consultar y considerar atentamente las recomendaciones y GPC actualizadas emitidas por autoridades sanitarias competentes. Es también responsabilidad del profesional verificar la normativa y la legislación sobre fármacos y dispositivos médicos a la hora de prescribirlos.
Las declaraciones de conflictoS de intereses de todos los expertos que han participado en la redacción de esta guía están disponibles en la página web de la ESC: www.escardio.org/guidelines.
9APÉNDICEFiliaciones de los autores y miembros del Grupo de Trabajo:
Sonya V. Babu-Narayan, NHLI, Imperial College London, Royal Brompton and Harefield NHS Foundation Trust, Londres, Reino Unido; Werner Budts, Congenital and Structural Cardiology, UZ Leuven and Department of Cardiovascular Sciences, KU Leuven, Lovaina, Bélgica; Massimo Chessa, ACHD Unit, Paediatric and Adult Congenital Heart Centre, IRCCS Policlinico San Donato, San Donato Milanese, Milán, Italia; Gerhard-Paul Diller, Department of Cardiology III, Adult Congenital and Valvular Heart Disease, University Hospital, Münster, Alemania; Bernard Iung, Cardiology, Bichat Hospital, París, Francia; Jolanda Kluin, Cardiothoracic Surgery, Amsterdam UMC, University of Amsterdam, Ámsterdam, Países Bajos; Irene M. Lang, Cardiology, Medical University of Vienna, Viena, Austria; Folkert Meijboom, Cardiology, University Medical Centre Utrecht, Utrecht, Países Bajos; Philip Moons, Department of Public Health and Primary Care, KU Leuven, Lovaina, Bélgica, e Institute of Health and Care Sciences, University of Gothenburg, Gotemburgo, Suecia, y Department of Paediatrics and Child Health, University of Cape Town, Ciudad del Cabo, Sudáfrica; Barbara J.M. Mulder, Cardiology, Amsterdam University Medical Center, Ámsterdam, Países Bajos; Erwin Oechslin, Medicine, Division of Cardiology, Peter Munk Cardiac Centre, University Health Network and University of Toronto, Toronto, Ontario, Canadá; Jolien W. Roos-Hesselink, Cardiology, Erasmus MC, Róterdam, Países Bajos; Markus Schwerzmann, Center for Congenital Heart Disease, Department of Cardiology, University Hospital Inselspital, University of Bern, Berna, Suiza; Lars Sondergaard, Cardiology, Rigshospitalet, Copenague, Dinamarca, y Katja Zeppenfeld, Leiden University Medical Centre, Lovaina, Países Bajos.
Comité de la ESC para la Práctica de las Guías (CPG): Stephan Windecker (Moderador) (Suiza), Victor Aboyans (Francia), Colin Baigent (Reino Unido), Jean-Philippe Collet (Francia), Veronica Dean (Francia), Victoria Delgado (Países Bajos), Donna Fitzsimons (Reino Unido), Chris P. Gale (Reino Unido), Diederick E. Grobbee (Países Bajos), Sigrun Halvorsen (Noruega), Gerhard Hindricks (Alemania), Bernard Iung (Francia), Peter Jüni (Canadá), Hugo A. Katus (Alemania), Ulf Landmesser (Alemania), Christophe Leclercq (Francia), Maddalena Lettino (Italia), Basil S. Lewis (Israel), Béla Merkely (Hungría), Christian Mueller (Suiza), Steffen E. Petersen (Reino Unido), Anna Sonia Petronio (Italia), Dimitrios J. Richter (Grecia), Marco Roffi (Suiza), Evgeny Shlyakhto (Federación Rusa), Iain A. Simpson (Reino Unido), Miguel Sousa-Uva (Portugal) y Rhian M. Touyz (Reino Unido).
Sociedades Nacionales de Cardiología de la ESC implicadas activamente en el proceso de revisión de la Guía ESC 2020 sobre el tratamiento de las cardiopatías congénitas del adulto
Alemania: German Cardiac Society, Claudia Walther; Argelia: Algerian Society of Cardiology, Naima Hammoudi; Armenia: Armenian Cardiologists Association, Svetlana V. Grigoryan; Austria: Austrian Society of Cardiology, Johannes Mair; Azerbaiyán: Azerbaijan Society of Cardiology, Galib Imanov; Bélgica: Belgian Society of Cardiology, Antoine Bondue; Bielorrusia: Belorussian Scientific Society of Cardiologists, Jouri Chesnov; Bosnia y Herzegovina: Association of Cardiologists of Bosnia and Herzegovina, Naser Nabil; Bulgaria: Bulgarian Society of Cardiology, Anna Kaneva; Chipre: Cyprus Society of Cardiology, Ourania Hadjisavva; Croacia: Croatian Cardiac Society, Margarita Brida; Dinamarca: Danish Society of Cardiology, Dorte Guldbrand Nielsen; Egipto: Egyptian Society of Cardiology, Maiy Hamdy El Sayed; Eslovaquia: Slovak Society of Cardiology, Iveta Simkova; Eslovenia: Slovenian Society of Cardiology, Katja Prokselj; España: Sociedad Española de Cardiología, Pastora Gallego; Estonia: Estonian Society of Cardiology, Raili Ermel; Federación Rusa: Russian Society of Cardiology, Olga Moiseeva; Finlandia: Finnish Cardiac Society, Juha Sinisalo; Francia: French Society of Cardiology, Jean-Benoit Thambo; Georgia: Georgian Society of Cardiology, Zviad Bakhutashvili; Grecia: Hellenic Society of Cardiology, George Giannakoulas; Hungría: Hungarian Society of Cardiology, Olga Hajnalka Bálint; Irlanda: Irish Cardiac Society, Christopher J. Lockhart; Israel: Israel Heart Society, Amiram Nir; Italia: Italian Federation of Cardiology, Adriano Murrone; Kirguistán: Kyrgyz Society of Cardiology, Olga Lunegova; Kosovo (República de): Kosovo Society of Cardiology, Artan Ahmeti; Letonia: Latvian Society of Cardiology, Ainars Rudzitis; Líbano: Lebanese Society of Cardiology, Zakhia Saliba; Lituania: Lithuanian Society of Cardiology, Lina Gumbiene; Luxemburgo: Luxembourg Society of Cardiology, Kerstin Wagner; Macedonia del Norte: North Macedonian Society of Cardiology, Elizabeta Srbinovska-Kostovska; Malta: Maltese Cardiac Society, Maryanne Caruana; Marruecos: Moroccan Society of Cardiology, Rachida Amri; Montenegro: Montenegro Society of Cardiology, Nebojsa Bulatovic; Noruega: Norwegian Society of Cardiology, Mette-Elise Estensen; Países Bajos: Netherlands Society of Cardiology, Berto J. Bouma; Polonia: Polish Cardiac Society, Lidia Tomkiewicz-Pajak; Reino Unido de Gran Bretaña e Irlanda del Norte: British Cardiovascular Society, Louise Coats; República Checa: Czech Society of Cardiology, Jana Rubackova-Popelova; Rumanía: Romanian Society of Cardiology, Ioan Mircea Coman; San Marino: San Marino Society of Cardiology, Marco Zavatta; Serbia: Cardiology Society of Serbia, Anastazija Stojsic-Milosavljevic; Suecia: Swedish Society of Cardiology, Bengt Johansson; Suiza: Swiss Society of Cardiology, Matthias Greutmann; Túnez: Tunisian Society of Cardiology and Cardio-Vascular Surgery, Essia Boughzela, y Ucrania: Ukrainian Association of Cardiology, Yuriy Sirenko.
.77.
En representación de la AEPC.
Grupo de Trabajo sobre el Tratamiento de Cardiopatías Congénitas del Adulto de la Sociedad Europea de Cardiología (ESC)
La lista de miembros del Comité de la Sociedad Europea de Cardiología (ESC) para la Elaboración de Guías de Práctica Clínica (GPC) y revisores del documento representantes de las Sociedades Nacionales de Cardiología se recoge en el apéndice.
Este artículo se publica y distribuye bajo los términos de Oxford University Press, Standard Journals Publication Model (https://academic.oup.com/journals/pages/open-access/funder-policies/chorus/standard-publication-model).