
La principal manifestación fenotípica estructural de la amiloidosis cardiaca es la hipertrofia del ventrículo izquierdo (HVI). La presencia de determinadas características asociadas con la HVI en las técnicas de imagen debe plantear otros diagnósticos diferenciales más allá de la amiloidosis en pacientes con HVI, incluso en los portadores de variantes patogénicas en el gen de la transtirretina (TTR).
Se presenta el caso de un varón de 60 años con antecedentes de hipertensión arterial, diabetes mellitus tipo 2 y tabaquismo. El paciente tenía antecedentes familiares de amiloidosis hereditaria por transtirretina (ATTRv), con varios familiares portadores de una variante de tipo missense (Val50Met) en el gen TTR. Además, su padre había sido diagnosticado de hemocromatosis.
Un cribado familiar efectuado en 2010 demostró que el paciente era portador de la variante patogénica familiar en TTR. El estudio inicial con electromiograma, el ecocardiograma, la resonancia magnética cardiaca (RMC) y la gammagrafía cardiaca (GGC) no mostraron hallazgos que indicaran amiloidosis cardiaca.
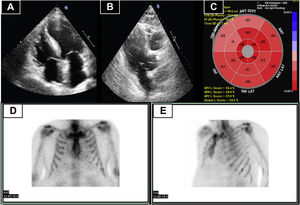
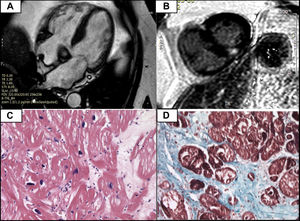
Cinco años después, el paciente tuvo fibrilación auricular. Ante esto, se decidió descartar ATTRv nuevamente. En ese momento el paciente se encontraba asintomático y la analítica mostraba cifras de la fracción aminoterminal del propéptido natriurético cerebral (NT-proBNP) levemente elevadas. El ecocardiograma (figura 1A-C) mostraba una función sistólica normal y una hipertrofia asimétrica a nivel septal basal, sin gradiente obstructivo. La deformación del corazón o strain longitudinal global estaba conservada y no mostraba un patrón típico de amiloidosis. La GGC no demostró captación miocárdica (figura 1D y E). En este caso, la HVI planteaba el diagnóstico diferencial entre amiloidosis cardiaca, hemocromatosis con afección cardiaca, cardiopatía hipertensiva y otras causas, por lo que se solicitó una RMC que confirmó la hipertrofia asimétrica con máximo grosor septal basal de 18 mm. En cuanto a la caracterización tisular, se obtuvo un valor de T2 normal. Además, se observó un realce tardío medioventricular focal en los puntos de inserción superior e inferior del ventrículo derecho (figura 2A y B).
. ANT: anterior; ANT-LAT: anterolateral; ANT-SEPT: anteroseptal; INF: inferior; INF-LAT: inferolateral; INF-SEPT: inferoseptal.")
A y B: ecocardiograma transtorácico que muestra un engrosamiento septal basal. C: patrón de strain longitudinal global del ventrículo izquierdo, con reducción de valores sobre todo a nivel anteroseptal e inferoseptal basal y medio. D y E: gammagrafía cardiaca que demuestra ausencia de captación cardiaca (puntuación de Perugini, 0). ANT: anterior; ANT-LAT: anterolateral; ANT-SEPT: anteroseptal; INF: inferior; INF-LAT: inferolateral; INF-SEPT: inferoseptal.
. C: ausencia de depósitos de amiloide por hematoxilina-eosina y rojo Congo. D: fibrosis por tinción tricrómica de Masson.")
A: resonancia magnética cardiaca que muestra hipertrofia septal asimétrica a nivel basal. B: presencia de realce tardío de gadolinio en los puntos de inserción del ventrículo derecho (típico de miocardiopatía hipertrófica de base sarcomérica, aunque no específico de esta). C: ausencia de depósitos de amiloide por hematoxilina-eosina y rojo Congo. D: fibrosis por tinción tricrómica de Masson.
A pesar de que los hallazgos de las pruebas de imagen no concordaban con el diagnóstico de amiloidosis cardiaca, ante la posibilidad de un falso negativo en la gammagrafía por la mutación Val50Met, se decidió llevar a cabo una biopsia endomiocárdica, que solo demostró fibrosis, sin infiltración amiloide (figura 2C y D).
Dado el patrón de strain longitudinal global en el ecocardiograma y de realce tardío en la RMC, así como la ausencia de amiloide en la biopsia endomiocárdica, se solicitó un nuevo estudio genético con panel next-generation sequencing de miocardiopatía hipertrófica (MCH). El análisis genético demostró la presencia de una variante patogénica en heterocigosis tipo missense (p.Gly771Ala) en el gen de la cadena pesada de la betamiosina (MYH7), además de la ya conocida en el gen TTR. Con ello, se estableció el diagnóstico de MCH sarcomérica no obstructiva, además de que el paciente era portador de una mutación en TTR sin afección cardiaca hasta ese momento.
Durante el seguimiento, el paciente contrajo una disfunción sinusal que condicionó el implante de un marcapasos. Ante la posibilidad de que se tratara de la primera manifestación de ATTRv por infiltración del sistema de conducción, se repitió la GGC 10 años después de la anterior. Se documentó una captación cardiaca de grado 1 de Perugini, y no se pudo establecer el diagnóstico no invasivo de ATTRv.
Dos años más tarde, una nueva GGC de control mostró una progresión de la captación, que alcanzó el grado 2 de Perugini. Esto, junto con la mutación en TTR, permitió establecer el diagnóstico de doble enfermedad en este paciente: MCH sarcomérica y ATTRv. El paciente dio su consentimiento informado para la publicación del caso.
El diagnóstico de amiloidosis cardiaca por transtirretina (ATTR) se puede obtener de manera invasiva o no invasiva1. Actualmente, una captación de grado 2/3 de Perugini en la GGC junto con el cribado de una discrasia de células plasmáticas permite diagnosticar a pacientes con ATTR de forma no invasiva con una elevada especificidad2. Una vez diagnosticada la ATTR, se debe hacer un estudio genético, por sus implicaciones terapéuticas y familiares1.
La GGC es una técnica muy sensible y específica en el diagnóstico de ATTR. Pese a ello, hay que tener presente que puede arrojar falsos positivos, como las formas AL, y falsos negativos1.
Clásicamente, se había descrito el patrón de hipertrofia concéntrica asociado con la amiloidosis por el carácter infiltrativo de la enfermedad; no obstante, el estudio más grande de pacientes con ATTR sometidos a RMC demostró que el patrón más habitual es el del septo sigmoide2. En cuanto a la caracterización tisular por RMC, un realce tardío de gadolinio subendocárdico general o transmural se considera diagnóstico de la enfermedad, junto con la alteración de la cinética del gadolinio1. Además, se suele acompañar de un valor en T1 nativo Elevado y valores muy altos de volumen extracelular3.
La combinación de una historia clínica que incluya banderas rojas, los antecedentes familiares, el electrocardiograma, las técnicas de imagen, la histología y el estudio genético son los medios disponibles para conseguir el diagnóstico certero de los pacientes con HVI.
DECLARACIÓNEl presente caso fue seleccionado para su publicación en Revista Española de Cardiología de entre los recibidos en la edición de 2023 de la Liga de los Casos Clínicos de la Sociedad Española de Cardiología.
CONTRIBUCIÓN DE LOS AUTORESEscritura y diseño del artículo y las figuras: D. de Castro. Revisión, edición del artículo y las figuras: E. González-López. Revisión del artículo: B. Angulo-Lara, D. Pujol-Pocull y C. Collado-Macián.
FINANCIACIÓNNo existe ninguna fuente de financiación asociada a esta investigación.
CONFLICTO DE INTERESESNinguno.