
Left ventricular hypertrophy (LVH) is the main structural phenotypic manifestation of cardiac amyloidosis. However, the presence of specific LVH-associated characteristics in imaging studies should suggest differential diagnoses beyond amyloidosis in patients with LVH, even in carriers of pathogenic variants in the transthyretin (TTR) gene.
We present the case of a 60-year-old man with a history of hypertension, type 2 diabetes mellitus, and smoking. The patient had a family history of hereditary transthyretin amyloidosis (ATTRv), with several family members carrying a missense mutation (Val50Met) in the TTR gene. In addition, the patient's father had been diagnosed with hemochromatosis.
Family screening conducted in 2010 revealed that the patient was a carrier of the pathogenic familial variant in TTR. The initial workup with electromyography, echocardiography, cardiac magnetic resonance imaging (CMR), and myocardial perfusion imaging (MPI) found no indicators of cardiac amyloidosis.
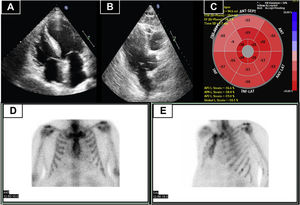
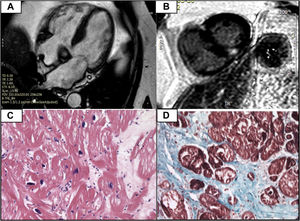
Five years later, the patient developed atrial fibrillation. In response, ATTRv was again ruled out. At this time, the patient was asymptomatic and blood tests revealed slightly elevated levels of N-terminal pro-B type natriuretic peptide (NT-proBNP). Echocardiography (figure 1A-C) demonstrated normal systolic function and asymmetric basal septal hypertrophy, without an obstructive gradient. Global longitudinal strain was preserved and showed no pattern typical of amyloidosis. MPI identified no myocardial uptake (figure 1D, E). At this time, the presence of LVH suggested differential diagnosis between cardiac amyloidosis, hemochromatosis with cardiac involvement, hypertensive heart disease, and other causes. Accordingly, CMR was performed, which confirmed the asymmetric hypertrophy, identifying a maximum basal septal thickness of 18 mm. Tissue characterization showed a normal T2 value. In addition, focal midventricular late enhancement was seen at the upper and lower right ventricular insertion points (figure 2A, B).
. ANT, anterior; ANT-LAT, anterolateral; ANT-SEPT, anteroseptal; INF, inferior; INF-LAT, inferolateral; INF-SEPT, inferoseptal.")
A and B: transthoracic echocardiography showing basal septal thickening. C: global longitudinal strain pattern of the left ventricle, with reduced values, particularly in the basal and mid-anteroseptum and inferoseptum. D and E: myocardial perfusion imaging showing absence of cardiac uptake (Perugini score, 0). ANT, anterior; ANT-LAT, anterolateral; ANT-SEPT, anteroseptal; INF, inferior; INF-LAT, inferolateral; INF-SEPT, inferoseptal.
. C: absence of amyloid deposits with hematoxylin and eosin and Congo red. D: fibrosis shown using Masson")
A: cardiac magnetic resonance imaging showing asymmetric septal hypertrophy at the basal level. B: presence of late gadolinium enhancement at the right ventricular insertion points (typical of sarcomeric hypertrophic cardiomyopathy, although not specific for it). C: absence of amyloid deposits with hematoxylin and eosin and Congo red. D: fibrosis shown using Masson's trichrome stain.
Although the imaging findings did not support a diagnosis of cardiac amyloidosis, we decided to conduct an endomyocardial biopsy, given the possibility of false-negative MPI results due to the Val50Met mutation. The results showed fibrosis, without amyloid infiltration (figure 2C, D).
Due to the global longitudinal strain pattern on the echocardiogram and the late enhancement on the CMR, as well as the absence of amyloid in the endomyocardial biopsy, a new genetic study was conducted with a next-generation sequencing panel for hypertrophic cardiomyopathy (HCM). The genetic analysis revealed the presence of a pathogenic heterozygous missense variant (p.Gly771Ala) in the gene for beta-myosin heavy chain (MYH7), in addition to the previously identified mutation in the TTR gene. Accordingly, the patient was diagnosed with nonobstructive sarcomeric HCM, as well as being a carrier of a mutation in TTR that, thus far, had no cardiac involvement.
During follow-up, the patient developed sinus node dysfunction requiring pacemaker implantation. Because this condition might have been the first manifestation of ATTRv, caused by amyloid infiltration of the conduction system, another MPI was performed, 10 years after the first scan. Perugini grade 1 cardiac uptake was detected, and a diagnosis of noninvasive ATTRv could not be established.
Two years later, repeat MPI showed uptake progression, which was now Perugini grade 2. This, together with the TTR mutation, led to a dual diagnosis in this patient: sarcomeric HCM and ATTRv. The patient gave his informed consent for publication of the case.
Transthyretin cardiac amyloidosis (ATTR) can be diagnosed invasively or noninvasively.1 Currently, Perugini grade 2/3 uptake on MPI together with screening for plasma cell dyscrasia allows noninvasive and highly specific diagnosis of patients with ATTR.2 Once ATTR is diagnosed, a genetic study must be performed due to its therapeutic and familial implications.1
MPI is a highly sensitive and specific technique for ATTR diagnosis. Nonetheless, there is always the possibility of false positives, such as the light-chain amyloidosis (AL) forms, and false negatives.1
Classically, the pattern of concentric hypertrophy associated with amyloidosis has been associated with the infiltrative nature of the disease. Nonetheless, the largest study of patients with ATTR who underwent CMR showed that the most common pattern is that of a sigmoid septum.2 For CMR-mediated tissue characterization, general or transmural subendocardial late gadolinium enhancement is considered diagnostic for the condition, together with alteration of the gadolinium kinetics.1 In addition, it is usually accompanied by an elevated native T1 value and very high extracellular volumes.3
The accurate diagnosis of patients with LVH relies on a combination of a clinical history including red flags, family history, electrocardiography, imaging studies, histology, and a genetic study.
DECLARATIONThe present case was selected for publication in Revista Española de Cardiología among those submitted to the 2023 edition of the League of Clinical Cases of the Spanish Society of Cardiology.
FUNDINGNo funding source is associated with this research.
AUTHORS’ CONTRIBUTIONSDrafting and design of the article and figures: D. de Castro. Revision, article editing, and figures: E. González-López. Article revision: B. Angulo-Lara, D. Pujol-Pocull, and C. Collado-Macián.
CONFLICTS OF INTERESTNone.