Palabras clave
INTRODUCCION
La distrofia miotónica es la enfermedad hereditaria del sistema neuromuscular más común en adultos. Se trata de un trastorno multisistémico que se hereda de forma autosómica dominante con penetrancia casi completa y con compromiso panmuscular, músculo esquelético, músculo cardíaco y músculo liso, aunque las manifestaciones clínicas más llamativas son la debilidad distal y la miotonía. La miotonía es un signo clínico fácil de identificar y se describe como la incapacidad para relajar la musculatura estriada tras su estimulación, por ejemplo, incapacidad para soltar la mano cuando se le estrecha al paciente. Se asocian, además, cataratas, calvicie precoz, alteraciones endocrinas y, a veces, cierto retraso intelectual. Las enzimas musculares están discretamente elevadas. Es una de las enfermedades producidas por expansión de tripletes, en este caso la expansión de un triplete CTG en un gen del cromosoma 19 que codifica una proteincinasa (miotonina). Como en la mayoría de las enfermedades producidas por expansión de tripletes, el número de repeticiones condiciona la edad de aparición y la gravedad de los síntomas.
La forma del adulto comienza entre los 15 y los 45 años y existe una forma congénita con síntomas desde el nacimiento. La afección cardíaca en estos pacientes es frecuente, fundamentalmente las alteraciones del ritmo y la conducción auriculoventricular, y rara vez producen insuficiencia cardíaca.
CASO CLÍNICO
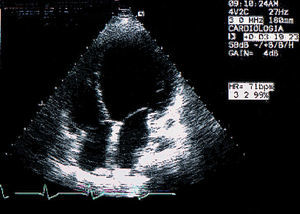
Varón de 36 años de edad, diagnosticado a los 20 años de distrofia miotónica de Steinert tras haber presentado dificultad progresiva para la deambulación, el habla y la masticación, así como debilidad muscular distal. Presentaba antecedentes familiares de distrofia miotónica (madre y tío materno fallecidos, afectados de distrofia miotónica sin afección cardiovascular conocida). El paciente había sido fumador de 15 paquetes/año, sin presentar criterios clínicos de bronquitis crónica, como único factor de riesgo cardiovascular. Consultó en nuestro hospital por disnea de esfuerzo progresiva de 2 meses de evolución, ortopnea y episodios de disnea paroxística nocturna, siendo ingresado con el diagnóstico de insuficiencia cardíaca de inicio. El paciente no refería clínica de angina ni datos sugestivos de miocarditis o cardiopatía isquémica, ni tampoco otros antecedentes de cardiopatía. En la exploración física presentaba una presión arterial de 120/70 mmHg y se encontraba afebril; la auscultación cardíaca era rítmica, con un tercer ruido, un soplo holosistólico mitral IV/VI, ingurgitación yugular de 10 cm a 45° y crepitantes bilaterales hasta tercio medio de ambos campos pulmonares. En la radiografía de tórax se apreciaba aumento del índice cardiotorácico, redistribución vascular y patrón intersticial bilateral. El electrocardiograma ponía de manifiesto un ritmo sinusal y una imagen de bloqueo completo de rama izquierda. En el ecocardiograma (fig. 1) se apreciaba un ventrículo izquierdo dilatado con hipocinesia generalizada y función ventricular severamente deprimida, así como una regurgitación mitral severa y una insuficiencia tricuspídea moderada que permitía medir una presión sistólica en la arteria pulmonar de 37 mmHg. En la analítica presentaba hemograma y coagulación normales, una bioquímica general sin alteraciones significativas con determinaciones de calcio, fósforo, ferritina, ácido fólico, vitamina B12 y potasio normales. Las serologías de la hepatitis y el VIH fueron negativas. Tras realizar tratamiento deplectivo la respuesta clínica fue satisfactoria, por lo que fue dado de alta en tratamiento con vasodilatadores, diuréticos a dosis bajas y anticoagulantes orales. Tres meses después permanecía asintomático, en buen grado funcional.

Fig. 1. Ecocardiograma bidimensional, eje apical-cuatro cámaras en que se observa una dilatación desproporcionada del ventrículo izquierdo, con válvulas mitral y tricúspide sin anomalías estructurales.
DISCUSIÓN
En la distrofia miotónica el corazón se ve afectado con frecuencia, aunque la mayoría de los pacientes no presenta síntomas cardíacos1. No existe una correlación entre la gravedad de la afección muscular esquelética y la cardíaca y resulta impredecible la edad de comienzo y la progresión de la afección cardíaca. El indicador más sensible de ésta es el electrocardiograma, ya que la frecuencia de las alteraciones electrocardiográficas oscila entre el 85 y el 90%, y predominan las alteraciones del sistema de conducción de His-Purkinje, como defectos de la conducción auriculoventricular e intraventricular2, bloqueo de rama derecha, bloqueo fascicular izquierdo y bloqueo auriculoventricular. A pesar de su elevada incidencia, la mayoría de las alteraciones de la conducción y del ritmo de estos enfermos son leves y, por tanto, subclínicas.
Globalmente, las arritmias auriculares o ventriculares se presentan hasta en el 50% de los pacientes, pero las arritmias ventriculares graves son poco frecuentes y de manera excepcional son la forma de presentación cardiológica de la enfermedad3. Por el contrario, en contraste con la alta prevalencia de las alteraciones electrocardiográficas, hay una baja frecuencia de pacientes sintomáticos desde el punto de vista cardíaco. Esto podría deberse a que la afección del músculo cardíaco, generalmente oculta, toma la forma de distrofia más que de miotonía no selectiva, con una distribución en apariencia igual en las cuatro cavidades del corazón, siendo rara la afección extensa del miocardio en grado suficiente como para dar origen a signos o síntomas clínicamente evidentes4. La asociación de disfunción ventricular importante puede incrementar el riesgo de arritmias ventriculares y muerte súbita de estos enfermos.
Así, menos del 10% de los pacientes tiene datos clínicos de insuficiencia cardíaca1. A pesar de ello, el ecocardiograma o la resonancia magnética revelan alteraciones en porcentajes que oscilan desde el 33 al 78% según las series5. Se ha descrito hipertrofia ventricular izquierda y derecha, dilatación ventricular derecha, disminución de la fracción de eyección del ventrículo izquierdo, infiltración grasa y fibrosis en el ventrículo derecho o biventricular y prolapso de la válvula mitral5. En las miopatías ligadas al cromosoma X, como las distrofias musculares de Duchene y Becker, suele presentarse miocardiopatía dilatada. Sin embargo, ésta es rara en la distrofia miotónica, como en el presente caso, forma inhabitual de presentación de síntomas cardiológicos en ausencia de trastornos del ritmo subyacente6. También se ha descrito el caso de fallo cardíaco y miopatía como complicación de la distrofia miotónica durante el embarazo7, tras descartar mediante biopsia endomiocárdica otras posibles causas de miocardiopatía potencialmente tratable, como la miocarditis inflamatoria o la miocardiopatía idiopática periparto.
La afección miocárdica indica la expresión completa de los genes del tejido muscular estriado, tanto esquelético como miocárdico. Por otro lado, dado que los tejidos de conducción especializados y el miocardio tienen orígenes embrionarios muy estrechos, si no idénticos, no es sorprendente que el marcador genético afecte a ambos.
Como limitaciones debe referirse que no es posible descartar de forma absoluta la posibilidad de una miocardiopatía dilatada idiopática, con lo que asistiríamos a la asociación de 2 enfermedades distintas. Con el presente caso exponemos una forma de presentación inhabitual de insuficiencia cardíaca en los pacientes con distrofia miotónica de Steinert6,7. Sin embargo, creemos que debe tenerse en cuenta como diagnóstico diferencial en pacientes entre la tercera y cuarta décadas de vida, cuya manifestación inicial es una insuficiencia cardíaca, en los que se ha realizado un cribado de las causas más prevalentes en este perfil de edad.
Al ser el diagnóstico de esta enfermedad eminentemente clínico (fenómeno miotónico) es necesario un alto índice de sospecha para poder realizar su diagnóstico si no existe un conocimiento previo de la enfermedad.
Correspondencia: Dr. F. Cabrera Bueno. Urbanización Coto de Rosas, fase 2, 14. 29620 Torremolinos. Málaga. Correo electrónico: fcabrerab@meditex.es Recibido el 10 de julio del 2000. Aceptado para su publicación el 30 de julio del 2000.