Palabras clave
INTRODUCCIÓN
El origen de la miocardiopatía hipertrófica (MCH) ha sido un enigma que ha fascinado a varias generaciones de cardiólogos desde que la describió Teare el año 19581. Aunque se conocía su carácter hereditario1, no era fácil explicar el mecanismo de sus alteraciones anatómicas: a) la hipertrofia (que solía atribuirse a una hiperactividad simpática congénita, entre otras hipótesis); b) la desorganización de los miocitos, y c) las lesiones de los pequeños vasos, o la patogenia de sus correspondientes manifestaciones clínicas: a) la disfunción mecánica sistólica y diastólica; b) la muerte súbita, y c) la isquemia miocárdica2,3. La solución ha tenido que esperar a las grandes contribuciones de la biología molecular de la última década.
MUTACIONES CAUSALES Y GRAVEDAD DEL FENOTIPO
El enigma empezó a desvelarse en 1990 con el descubrimiento, en el laboratorio de la Dra. Seidman, de que la causa era una mutación puntual (missense mutation) de la cadena pesada de la β-miosina (β-MHC [myosin heavy chain]), la principal proteína contráctil del sarcómero4. Basta un cambio de un nucleótido entre los 23.000 pares de bases que componen el gen (MYC7) que codifica la molécula para provocar la enfermedad.
Desde entonces se han descrito numerosas mutaciones causales (más de 150), no sólo en el gen de la miosina sino también en el resto de las proteínas que componen el aparato contráctil (por lo que se la llama la enfermedad del sarcómero) (fig. 1). Las mutaciones pueden afectar a las proteínas motoras propiamente dichas, las moléculas reguladoras de la troponina I y T (TnI y TnT), α-tropomiosina; o las proteínas estructurales, como las proteínas gigantes titina, que mantienen unidas las fibras en sentido axial con el disco Z y la proteína C de unión a la miosina (MyBPC [myosin binding protein C]) que las une en sentido transversal. En la tabla 1 se resumen algunas características de los 9 genes causales2,5-7.

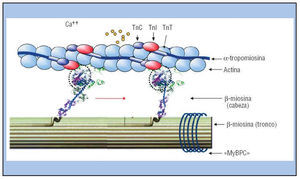
Fig. 1. Esquema de las proteínas contráctiles del sarcómero. Muestra el filamento delgado de actina (doble collar de perlas) con el complejo tropomiosina-troponina dispuesto a lo largo de su surco (troponinas T [TnT], I [TnI] y C [TnC] y α-tropomiosina), que inhibe la interacción de la cabeza de la miosina con la actina. El filamento grueso está formado por un haz de ≈ 400 moléculas de β-miosina (unidas por la proteína C de unión a la miosina [MyBPC]), cuyas cabezas motoras (2 por molécula) interaccionan con la actina, en el momento en que el Ca++ se une a la TnC y libera la inhibición. La flecha roja indica el movimiento de flexión de la cabeza y el cuello (60°) que desplaza el filamento grueso.

La gran hetereogeneidad genética ofrece una primera explicación a la diversidad de manifestaciones clínicas que presenta la enfermedad (fenotipos), en términos de la importancia de la hipertrofia o la propensión a la muerte súbita y su pronóstico2,7-10. Aunque las correlaciones genotipo/fenotipo son todavía imprecisas, dado que el número de familias estudiadas es todavía limitado, pueden distinguirse de manera esquemática 3 formas clínicas: la forma clásica debida habitualmente a una mutación de la β-MHC, que da lugar a una hipertrofia casi siempre masiva, de predominio septal, que se detecta precozmente (en la pubertad) y tiene una tendencia notable a la muerte súbita; la que se asocia con la TnT, que tiene especial tendencia a la muerte súbita y escasa hipertrofia9, y las de la MyBPC, en las que la aparición de la hipertrofia es muy tardía y la evolución benigna10.
Ello explica que la sensibilidad diagnóstica del ecocardiograma (grosor del tabique o la pared libre del ventrículo izquierdo ≥ 13 mm) puede ser adecuada para la detección de hipertrofia en pacientes con mutaciones de la β-MHC, pero escasa en otras variantes de MCH, como la TnT y la MyBPC, en las que la hipertrofia es poco aparente o más tardía10.
Pero además, la gravedad de la hipertrofia y el pronóstico dependen no sólo del gen mutante, sino también de la localización de las mutaciones en determinadas zonas o dominios funcionales críticos de la molécula y de si la mutación modifica la polaridad eléctrica del aminoácido y, por consiguiente, la configuración (el plegamiento) de la proteína. Por lo que respecta a la β-miosina, la proteína mejor estudiada, las mutaciones causales conocidas se sitúan exclusivamente en la cabeza o motor molecular. Pueden estar en zonas con actividad bioquímica hidrolítica: la zona de contacto con la actina, en estrecha relación con la zona con actividad adenosina trifosfatasa (ATPasa) dependiente de la actina, en forma de hendidura donde se aloja la molécula de ATP, que al hidrolizarse libera un enlace fosfórico de alta energía (. P), necesario para la contracción. También pueden ocurrir en las zonas con actividad mecánica: las asas motoras causales del movimiento de torsión; la zona del «convertidor», que une las asas motoras al largo brazo de palanca (α-helix) que amplifica el movimiento y al rotar unos 60° da el golpe de remo que desliza la miosina sobre la actina (mientras la unión de la cabeza con la actina se mantiene firme y no sufre ninguna flexión)2,11-14 (fig. 2 y tabla 2).

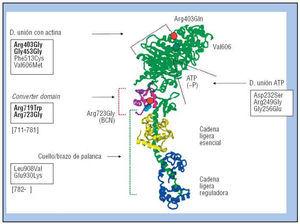
Fig. 2. Diagrama de cintas de la estructura de la cabeza motora/hidrolítica de la β-miosina, el motor molecular del corazón. En verde, la cabeza y el cuello o brazo de palanca (α-helix), unidos por la pequeña zona del convertidor (converter domain, en rojo-púrpura). Los círculos rojos señalan la localización de la mutación Arg403Gln en el borde de la zona de unión con la actina y de la Arg723Gly (mutación Barcelona) en el «convertidor», respectivamente. Adosadas al cuello o brazo de palanca (en verde), aparecen las cadenas ligeras esencial (en amarillo) y reguladora (en violeta). En recuadro, las mutaciones más conocidas y en negrita las más graves. (Adaptada de Köhler et al27. Cortesía de la Dra. T. Kraft.).

Incluso dentro de la misma familia con la misma mutación, el fenotipo puede variar en función de los llamados factores modificadores (estilo de vida, entre los que parece tener una importancia especial el ejercicio, o la hipertensión arterial), la coexistencia de otras mutaciones en el mismo gen (6% de los casos) o en otro gen (second hit) o la presencia de genes modificadores, como el polimorfismo I/D de la enzima de conversión de la angiotensina y el 1166 A/C del receptor de tipo 1 de la angiotensina 12,15.
MECANISMOS DE LA HIPERTROFIA
Para explicar el mecanismo de la hipertrofia se han propuesto 2 hipótesis, la extrínseca y la intrínseca, que no tienen por qué ser totalmente excluyentes (fig. 3).

Fig. 3. Hipótesis extrínseca e intrínseca. Para la explicación véase el texto.
Hipótesis extrínseca (extracelular). La hipertrofia adaptativa
La presencia de una mutación en una proteína contráctil sugería a priori que la hipertrofia era la respuesta compensadora destinada a aumentar la generación de fuerza para contrarrestar el déficit funcional15. El mecanismo no sería sustancialmente distinto del de la hipertrofia adaptativa de las sobrecargas mecánicas o del postinfarto de miocardio, que tienden a normalizar el estrés excesivo y aumentar la potencia para mantener el gasto cardíaco16,17. Los estímulos desencadenantes pueden ser la distensión mecánica del miocito (el estrés o estiramiento del sarcolema) y la activación neurohumoral simpática del sistema renina-angiotensina circulante o local, o de las citocinas. El fenotipo de la célula hipertrófica es similar: aumenta el tamaño del miocito, gracias al aumento de la síntesis de proteínas, con reinducción del «patrón fetal», que se reconoce por el aumento de la expresión de determinados genes, como los del péptido natriurético atrial, y la menor expresión de otros, como los correspondientes a los canales del calcio, por ejemplo18,19.
Pero los estudios experimentales que analizan la velocidad o la fuerza de contracción (los 2 parámetros inversamente relacionados entre sí que describen la potencia o actividad mecánica) no han podido demostrar de manera consistente ningún déficit funcional. Al contrario, la mayoría de los trabajos sugieren un aumento de la potencia de la fibra muscular mutante, lo que pone en duda la validez de esta hipótesis como mecanismo unitario de la MCH14,20.
Un buen ejemplo de esta controversia son los estudios relacionados con 2 de las 4 mutaciones más graves de la β-MHC (tabla 2)2: la Arg403Gln, la primera conocida, que está situada en una zona con actividad bioquímica y afecta primariamente a la cinética o la velocidad de la contracción; y la Arg703Gly (mutación Barcelona) descrita por nosotros21, que afecta a la zona del convertidor (converter domain), una zona que desempeña un papel crítico en la actividad mecánica de la cabeza de la miosina.
Alteraciones funcionales de la mutación Arg403Gln de la β-MHC
Mediante la prueba de la motilidad in vitro, que permite observar al microscopio el movimiento de las moléculas o los filamentos aislados de actina fluorescente sobre una capa de cabezas de miosina mutada, adheridas a un porta de cristal, Cuda et al22 describieron una importante disminución de la velocidad máxima de desplazamiento de la actina (Vmáx), un parámetro que se correlaciona con la actividad ATPasa dependiente de la actina. Las moléculas de β-miosina mutada se obtuvieron a partir de biopsias de músculo esquelético de pacientes afectados de MCH. Al igual que los miocardiocitos, los miocitos coexpresan proteína mutante y proteína normal ya que la β-miosina de las fibras musculares lentas (tipo I) está codificada por el mismo gen que en el miocardio. La proporción relativa de la miosina mutante varía por lo general entre el 30 y el 50%.
La presencia de un déficit funcional de miosina parecía reforzar la hipótesis de la hipertrofia compensadora. Sin embargo, los investigadores del laboratorio de la Dra. Seidman, utilizando ratones transgénicos que expresan la mutación Arg403 (α-MHC403/+) y reproducen con gran fidelidad la MCH humana, detectaron un aumento de la motilidad de la actina in vitro (la Vmáx aumenta en un 60%) y de la actividad enzimática ATPasa23,24. Y por si había dudas acerca de la validez del modelo del ratón, cuya miosina es α-miosina o miosina rápida (la frecuencia cardíaca es muy elevada), este aumento funcional se confirmó también en muestras de β-miosina humana (miosina lenta) obtenida a partir de una biopsia cardíaca en pacientes con MCH25.
Además, los estudios hemodinámicos realizados en el ratón transgénico con registro simultáneo de las curvas de presión (con micromanómetro) y de volumen ventricular (mediante catéter de volumen) confirmaron que la mutación se asocia a un aumento significativo de la contractilidad (aumento de la dP/dT y de la pendiente de la relación presión/volumen telesistólica en el diagrama de presión/volumen). Comprobaron también que el defecto funcional más destacado y temprano del miocardio es el enlentecimiento de la relajación (dP/dT negativo), que aparece antes de que se manifieste la hipertrofia. Esta alteración tiene carácter progresivo, como confirman los registros a las 6 y a las 20 semanas. Estos hallazgos subrayan de nuevo que la MCH es básicamente una enfermedad de la diástole26.
Mutaciones del dominio del convertidor. La mutación Arg723Gly de la β-MHC (mutación Barcelona)
Los estudios acerca de las propiedades contráctiles de la fibra muscular aislada procedentes de la biopsia del músculo esquelético de pacientes con la mutación Arg719Try, una mutación contigua a la Arg723Gly, demostraron también un incremento de la función, caracterizado en este caso por un aumento del 50-60% en la fuerza máxima de contracción isométrica (fig. 4), atribuible a la mayor capacidad contrácil de las cabezas motoras de la miosina. La Vmáx de acortamiento, parámetro sensible a la cinética de la interacción actina-miosina, no se afecta por la mutación27.

Fig. 4. Efecto de la mutación Arg719Trp en la función mecánica de las fibras musculares aisladas procedentes del músculo gemelo. Las gráficas muestran un aumento del 50-60% de la fuerza generada y de la rigidez de la fibra en condiciones isométricas. La velocidad máxima de acortamiento (Vmáx) no se modifica (Adaptada de Köhler et al27. Cortesía de la Dra. T. Kraft.)
¿Cómo puede explicarse este aumento aparentemente paradójico de la fuerza contráctil? La investigación de las consecuencias funcionales de la mutación Arg723Gly, realizada en colaboración con T. Kraft y B. Brenner, del laboratorio de Fisiología Molecular y Celular de la Facultad de Medicina de Hannover (Alemania), ha permitido aportar nueva luz en este tema. Los resultados preliminares del estudio sobre la fibra muscular aislada (skinned fiber), obtenida a partir de la biopsia de músculo gemelo, han revelado una reducción de la sensibilidad del calcio, a juzgar por el desplazamiento de la curva de fuerza-pCa a la derecha en relación con la curva obtenida en las fibras de control (fig. 5). Las fibras que contienen la mutación, a la misma concentración de calcio generan menos fuerza, por lo que es necesario aumentar la concentración de calcio para alcanzar el mismo nivel28. Esta disminución de la sensibilidad al calcio, de origen incierto, es un dato completamente nuevo en las mutaciones de la miosina y puede considerarse la prueba definitiva de que existe un defecto funcional de la cabeza motora que afecta a las propiedades contráctiles básicas de la fibra muscular. Esto permite inferir que el aumento de la fuerza contráctil de la fibra muscular no es la alteración primaria, sino que es ya un mecanismo compensador.

Fig. 5. Efecto de la mutación Arg723Gly (mutación Barcelona) en la sensibilidad al calcio de las fibras musculares aisladas (curvas de fuerza/concentración de Ca++ [pCa]), procedentes de una biopsia de músculo gemelo. La sensibilidad al calcio está disminuida como indica el desplazamiento de la curva a la derecha en relación con los controles normales. A pCa 6,27 (línea de puntos) las fibras que contienen la mutación (cuadros negros) generan un 30% menos de fuerza que las fibras de control (círculos en blanco). T, 20 °C; n = 13 fibras, ± EEM (error estándar de la media). (Cortesía de la Dra. T. Kraft.)28
Pero ¿cuál puede ser el defecto capaz de exigir un aumento de la capacidad contráctil?
Los estudios en fibra aislada habían demostrado ya que las mutaciones que afectan al convertidor (Arg719Trp) dan lugar a un aumento de la rigidez de la fibra muscular atribuible a un aumento de la resistencia de la cabeza de la miosina a la distorsión elástica. La mutación es un excelente marcador que permite localizar esta resistencia, con toda precisión, en el dominio del convertidor, que se reafirma por consiguiente como el elemento elástico o flexible de la cabeza, que transmite la torsión de las asas motoras de la cabeza globular al brazo de la palanca (el resto de la cabeza es relativamente rígido)27. Asumiendo que el defecto funcional de la Arg723 es idéntico al de la Arg719, cabe deducir que el aumento de la fuerza de la contracción isométrica representa el sobresfuerzo necesario para vencer la mayor resistencia de las cabezas de la miosina mutada a la flexión (pueden ser mutantes la mitad de las cabezas de la miosina), y esta compensación se alcanza probablemente a expensas de un aumento del calcio disponible en el sarcómero, lo que nos acerca a la hipótesis intrínseca.
Por otra parte, la disminución de la sensibilidad al Ca y la disparidad funcional con los miocitos vecinos podrían ser otra causa de la disfunción contráctil28.
Hipótesis intrínseca (intracelular). Hipertrofia maladaptativa. Alteración de la movilización del Ca++ mioplasmático
Según la hipótesis intracelular, la hipertrofia es un efecto colateral del aumento del calcio en el aparato contráctil, que sería la respuesta compensadora primaria destinada a potenciar la generación de fuerza19,29. En último término la capacidad contráctil de los miofilamentos depende del calcio disponible localmente (y de la sensibilidad al Ca de las proteínas reguladoras). Pero el calcio desempeña un doble papel fisiológico en el miocito: aparte de que aumenta la contractilidad, es también un poderoso estímulo que activa la cascada de señales intracelulares que potencian la síntesis de proteínas y la hipertrofia (y en particular la vía calcineurina-calmodulina). La hipertrofia parece un elemento compensador imprescindible y podría tener en cambio consecuencias desfavorables (hipertrofia maladaptativa)29-31.
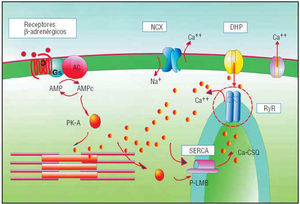
Estudios recientes han descrito una serie de alteraciones de la movilización del calcio intracelular32 que avalan esta hipótesis. Cabe recordar que la magnitud de la onda de calcio (el calcium transient) que estimula la contracción (y la síntesis de las proteínas) se regula por un complejo mecanismo en el que intervienen (fig. 6): a) los canales del calcio tipo L dependientes del voltaje (los receptores dihidropiridínicos), que al abrirse por efecto del potencial de acción permiten la entrada de una cantidad mínima de calcio en el citosol; b) los canales de la rianodina (RyR), que responden a dicho estímulo liberando de manera explosiva calcio del sistema retículo-endoplasmático (SERCA) que dispara la contracción, y c) los canales de calcio dependientes de la ATPasa (SERCA), la bomba de calcio de la membrana del SRE, que recaptan de manera masiva el calcio citosólico y determinan la relajación29.

Fig. 6.Esquema de las proteínas y los canales que regulan la magnitud de la onda de calcio. Las mutaciones del receptor de la rianodina (RyR) Arg929Cys y Glu3654Asp son una nueva causa de miocardiopatía hipertrófica que afecta a una proteína no contráctil35. CSQ: calsecuestrina (la proteína del interior del SRE que tiene gran afinidad por el calcio y facilita su depósito y su movilización); DHP: canal dihidropiridínico o canal L; NCX: intercambiador Na/Ca; P-LMB: fosfolambán; RyR: canal de rianodina; SERCA: bomba de calcio del sistema retículo-sarcoplasmático.
En el modelo de rata transgénica (α-MHC403/+) se ha comprobado una reducción notable de la expresión de los canales de la rianodina, del depósito de Ca++ y calsecuestrina del SRE, que los autores atribuyen a un atrapamiento del calcio en las fibras contráctiles que disminuye la disponibilidad de calcio en el resto de los compartimentos del mioplasma33.
Por otra parte, el estudio del metabolismo del miocardio en pacientes con MCH, mediante espectroscopia de resonancia magnética con P-31, ha confirmado la existencia de un déficit bioenergético del miocito, a juzgar por la disminución del 30% del cociente fosfocreatina/ATP, que aparece tanto en las mutaciones de la miosina como de la TnT cardíaca y de la MyBPC, con independencia de que exista o no hipertrofia. Este hallazgo sugiere que la contracción en presencia de proteínas defectuosas ocasionaría un aumento excesivo del consumo energético (de ATP) en detrimento de otras funciones como la de bomba de calcio dependiente de la ATP (SERCA) que consume mucha energía. Esta hipótesis del «compromiso energético» explicaría el defecto de la recaptación del calcio del sarcómero, el deterioro de la función diastólica y el descenso del calcio del SRE34.
Una elegante confirmación de la importancia de la movilización del calcio en la patogenia de la hipertrofia genética es el hallazgo reciente de mutaciones causales de la MCH en el gen del receptor de la rianodina cardíaca (fig. 6), localizadas por vez primera en una proteína no contráctil. Se encontró en 3 de las 63 familias estudiadas (el 5% de los casos), con hipertrofia severa, arritmias ventriculares y obstrucción subaórtica35.
MECANISMO DE LAS ARRITMIAS VENTRICULARES Y LA MUERTE SÚBITA
El origen de la inestabilidad eléctrica que predispone a la muerte súbita por fibrilación vcular, el episodio más dramático e imprevisible de la enfermedad, sigue siendo un tema polémico.
Desorganización miocelular
El sustrato arritmogénico se relacionó inicialmente con la pérdida del paralelismo normal de los miocitos que se observa en los cortes histológicos, que podría alterar las propiedades electrofisiológicas del sincitio miocárdico. Esta desorganización se consideró un hecho inherente al defecto genético de las proteínas contráctiles, que dificultaría el ensamblaje normal de los miofilamentos y los miocitos, pero no es una alteración constante36.
Hutchins y Bulkley37 sugirieron hace años que la desorganización era un signo inespecífico de contracción isométrica del tabique interventricular. En ausencia de acortamiento faltaría el estímulo que orienta la disposición paralela de las fibras. Según estos autores, el tabique tiene tendencia a adoptar una morfología catenoide, una superficie con doble curvatura en silla de montar, generada por la revolución de una curva catenaria. La curva o arco catenario (el arco que introdujo Gaudí en la arquitectura) tiene la particularidad de que distribuye a la perfección las cargas que soporta (de ahí la ventaja del arco de Gaudí sobre el arco románico y el gótico). La contracción de esta superficie con 2 curvaturas catenarias opuestas produce acortamiento cero (isométrica), porque se contrarrestan perfectamente las fuerzas en los 2 planos.
Remodelado eléctrico. Desregulación del calcio y prolongación del potencial de acción
Hoy se concede gran importancia a la alteración de las propiedades electrofisiológicas inducidas por el llamado «remodelado eléctrico» que acompaña al remodelado de la hipertrofia. El remodelado modifica la expresión de los canales iónicos que regulan la repolarización de la membrana celular38-40 y provoca un desequilibrio entre las corrientes iónicas de entrada (a través del canal del calcio tipo L y del intercambiador sodio/calcio principalmente) y las de salida (canales del potasio) a favor de las primeras, lo que tiene como principal consecuencia la prolongación del potencial de acción, que predispone a la dispersión de los períodos refractarios y los mecanismos de reentrada. El cambio en la expresión de los canales iónicos puede atribuirse a la reexpresión del patrón fetal o a los mecanismos que alteran la movilización del calcio del mioplasma, agravados por la mayor heterogeneidad de la repolarización eléctrica (la mayor dispersión de los períodos refractarios) debida a la asimetría de la hipertrofia.
También pueden facilitar la aparición de arritmias ventriculares la presencia de isquemia, frecuente en la MCH y, sobre todo, la fibrosis del miocardio que favorece las microrreentradas, como ocurre en el infarto de miocardio. La fibrosis puede ser un componente importante de la hipertrofia (puede alcanzar el 20-40% de la masa miocárdica) y se interpreta como un fenómeno de sustitución de fibras musculares que han sufrido un proceso de apoptosis o necrosis por exceso de estimulación del calcio o por brotes recurrentes de isquemia de pequeños vasos. La relación de la fibrosis con la muerte súbita es compleja, porque también contribuye de manera decisiva al deterioro de la función diastólica: la rigidez del miocardio atribuible a la fibrosis se suma al defecto de la relajación causado por la insuficiente movilización del calcio. Se ha señalado que la disfunción diastólica crítica, en el momento de la taquicardia del esfuerzo, puede ser una causa mecánica de muerte súbita.
MECANISMO DE LA ISQUEMIA. RELACIÓN CON EL DEFECTO DE LA RELAJACIÓN
Las manifestaciones isquémicas son relativamente frecuentes en la MCH y pueden tener diversas causas:
Las alteraciones estructurales de las pequeñas arterias coronarias intramurales, caracterizadas por el engrosamiento fibroso de la íntima y la hipertrofia de la media, que pueden considerarse una forma de enfermedad de los pequeños vasos. Son capaces de reducir la reserva coronaria y provocar eventos de isquemia y necrosis focal, con fibrosis de sustitución41.
La hipertrofia masiva, cuya demanda excede la capacidad de la red coronaria39.
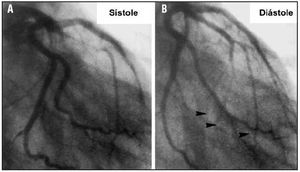
La compresión sistólica de los pequeños vasos intramurales (milking), que en principio no tiene por qué afectar a la circulación coronaria que tiene lugar en la diástole. Pero en presencia de una relajación diastólica muy lenta, la compresión puede prolongarse hasta bien avanzada la diástole y comprometer el riego coronario. Como pudimos confirmar mediante coronariografía en 5 pacientes con MCH y milking del 100% de la arteria coronaria descendente anterior, el tiempo que tarda en restablecerse el flujo en las ramas perforantes distales, una vez interrumpido el flujo en la arteria descendente anterior proximal, puede alcanzar al 20-33% de la diástole (fig. 7)42.

Fig. 7. Oclusión sistólica total de la arteria coronaria descendente anterior y de sus ramas septales en un paciente con miocardiopatía hipertrófica. A. En sístole. B. En diástole. El tiempo que tardan en rellenarse las ramas distales es de 140 ms, lo que equivale al 25% del período de llenado diastólico.
En apoyo de la relación de la isquemia con la disfunción diastólica, pudimos comprobar que los pacientes con MCH con angina tienen una relajación diastólica significativamente más lenta que los pacientes sin angina, a juzgar por la velocidad máxima de adelgazamiento de la pared libre del ventrículo izquierdo determinada mediante la angiografía cuantitativa (el equivalente al mean velocity gradient del Doppler tisular). El resto de los parámetros relacionados con la demanda (gravedad de la hipertrofia, índice presión/tiempo) no mostró ninguna diferencia apreciable (fig. 8).

Fig. 8. Relación de la angina con el defecto de la relajación miocárdica, en pacientes afectados de miocardiopatía hipertrófica. Los pacientes con angina muestran un enlentecimiento significativo de la velocidad máxima de la dilatación (dV/dt.V) y del adelgazamiento de la pared del ventrículo izquierdo (dh/dt.h) en la diástole precoz, determinadas en la ventriculografía cuantitativa (angiografía de contraste).
Este mecanismo puede agravarse en presencia de una arteria descendente anterior intramural (milking), anomalía del desarrollo que se presenta con relativa frecuencia en la MCH (y quizá forma parte de su cuadro clínico)41-43.
IMPLICACIONES CLÍNICAS
Diagnóstico ecocardiográfico
El hecho de que la hipertrofia no pueda considerarse el defecto sine qua non de la enfermedad, ya que su aparición puede ser muy tardía o inexistente en algunas mutaciones como la TnT y la MyBPC, impone ciertas limitaciones al diagnóstico basado en la medición del grosor ventricular. La identificación del defecto de relajación, que se ha revelado como la alteración más constante y temprana de la MCH, puede ser una buena alternativa, como sugieren 2 índices ecocardiográficos que se han propuesto recientemente:
La velocidad del movimiento del anillo mitral en dirección longitudinal, cuantificado mediante la ecografía Doppler tisular. Se ha visto que la disminución de las velocidades máxima del llenado precoz (Em) o tardío (Am) permite identificar los casos familiares que desarrollarán la hipertrofia en los siguientes 2 años44,45.
El gradiente máximo negativo de la velocidad del miocardio determinado mediante el Doppler tisular, que mide la velocidad del adelgazamiento diastólico de la pared ventricular y permite detectar el enlentecimiento de la relajación activa46. Este índice mide la diferencia entre la velocidad del movimiento del epicardio y el miocardio dividido por el espesor de la pared ventricular.
Diagnóstico genético
Dadas la diversidad y la falta de especificidad del fenotipo, y la posible coexistencia de otras causas, puede ser difícil tener la certeza del diagnóstico a menos que se confirme la mutación causal. Sin embargo, los métodos actuales del análisis genético son todavía muy engorrosos y de un coste muy elevado, por lo que su aplicación clínica está llena de dificultades. Además no son capaces de detectar más allá del 20-60% de las mutaciones. En un estudio reciente (EUROGENE Heart Failure Project) en el que se determinó la secuencia completa de 9 genes en 197 familias, el diagnóstico positivo se consiguió en el 63% de los casos7,47.
Pero el debate ya no es si debe o no intentarse el diagnóstico genético. El tema es saber cómo y cuándo las innovaciones técnicas harán factible su empleo habitual. La necesidad de la identificación de los portadores de la mutación se justifica ya por la posibilidad la instaurar precozmente un tratamiento preventivo de la muerte súbita, que en la actualidad significa muchas veces la implantación de un desfibrilador automático implantable2,7,47.
Intervención farmacológica temprana y prevención de la hipertrofia
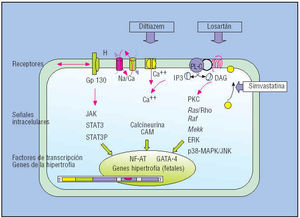
El tratamiento preventivo mediante una intervención farmacológica temprana, antes de que aparezca la hipertrofia, es un nuevo objetivo terapéutico que se sustenta en el concepto de que la hipertrofia es un efecto maladaptativo, perjudicial e innecesario para el mantenimiento de la función ventricular48. Aunque se ignora si las vías que utiliza la hipertrofia genética son las mismas que las de la hipertrofia adquirida, en principio parece posible la supresión de la hipertrofia mediante el bloqueo de alguna de las vías de señales intracelulares que controlan la expresión de los genes a nivel transcripcional, la síntesis de las proteínas contráctiles y el crecimiento de los miocitos (fig. 9), o el bloqueo de los receptores implicados: los canales del calcio, los receptores heptahelicoidales (ATII), y el receptor tipo interleucina16,17.

Fig. 9. Esquema de los receptores, las vías de señales intracelulares (cascadas de proteincinasas y fosfatasas) y los factores de transcripción que podrían estar implicados en la inducción de la hipertrofia. Los recuadros en blanco incluyen los fármacos que se han mostrado capaces de reducir la hipertrofia en el animal transgénico. Las flechas señalan su posible mecanismo de acción.
Los experimentos en modelos transgénicos son muy alentadores. En el ratón que expresa la mutación de la TnT Arg92Glu, la administración de losartán reduce la fibrosis, el colágeno y el factor de crecimiento transformador 149. También se ha comprobado el efecto beneficioso del diltiazem32, antagonista de los canales del calcio, que ya se emplea en clínica con el objetivo de mejorar la función diastólica. Y lo que es más sorprendente, se ha comprobado en el conejo transgénico MCH 403 que la simvastatina es capaz de reducir la hipertrofia del miocito y la fibrosis del miocardio, gracias a su efecto inhibidor de las señales intracelulares relacionadas con el sistema renina-angiotensina-aldosterona50. La simvastatina, aparte de que bloquea la síntesis del colesterol, también inhibe la síntesis de los isoprenoides, con lo que impide la activación (isoprenilación) de las pequeñas proteínas Rho y su traslocación a la membrana, donde es necesaria para que se materialice el estímulo de la hipertrofia en los miocitos o se active la proliferación de los fibroblastos (fig. 9).
El objetivo más ambicioso de la medicina preventiva, la terapia génica, queda más lejos, pero quizá menos de lo que parece. Teóricamente es posible anular la expresión del gen mutante si se bloquea la replicación del ARN mediante un oligonucleótido contrasentido. Dado que las proteínas del corazón humano se remplazan por completo en el término de pocas semanas, cabe la posibilidad de que las nuevas proteínas se formen sólo a partir del gen normal y desaparezca la hipertrofia51.
AGRADECIMIENTOS
A los profesores T. Kraft y B. Brenner y al Dr. A. Francisco por su inestimable colaboración en el estudio de la mutación BCN.
ABREVIATURAS
ATPasa: adenosina trifosfatasa.
β-MHC: cadena pesada de la β-miosina.
MCH: miocardiopatía hipertrófica.
MyBPC: proteína C de unión de la miosina.
SERCA: bomba de calcio del sistema retículo-sarcoplasmático.
TnC: troponina C.
TnI: troponina I.
TnT: troponina T.
Correspondencia: Prof. F. Navarro López.
Servicio de Cardiología (ICMCV). Hospital Clínic.
Villarroel, 179. 08036 Barcelona. España.
Correo electrónico: navarro@medicina.ub.es