El síndrome de Marfan (MIM 154700) es una enfermedad autosómica dominante con afección esquelética, ocular y cardiovascular, que tiene una prevalencia de 2-3/10.000 individuos. Según la nosología modificada de Gante, la presencia de una mutación patogénica en el gen de la fibrilina-1 (FBN1) asociada a dilatación de la raíz aórtica es suficiente para establecer su diagnóstico1. Se han identificado más de 1.800 mutaciones, la mayoría específicas de una sola familia, siendo el 25% de novo, sin haberse establecido una correlación entre genotipo y fenotipo por la variabilidad intrafamiliar e interfamiliar1. El mosaicismo parental podría explicar esta diferente expresión fenotípica y debería tenerse en cuenta en los casos de novo para el correcto consejo genético. Sin embargo, se han descrito pocas familias con síndrome de Marfan asociado a mosaicismo en FBN12–5.
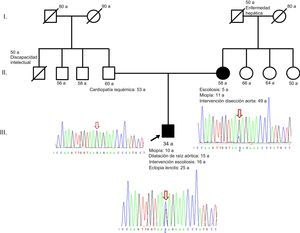
Describimos una nueva mutación en mosaicismo en FBN1que afecta al splicing. El probando es un varón de 34 años de edad, hijo único, diagnosticado de síndrome de Marfan por ectopia lentis y dilatación de la raíz aórtica, con una puntuación sistémica de 4 (miopía > 3 dioptrías, signo del pulgar, asimetría pectoral, escoliosis intervenida). Presenta los siguientes antecedentes familiares: padre con cardiopatía isquémica revascularizada y madre intervenida de disección aórtica tipo B con implante de endoprótesis aórtica percutánea (figura).
. El alelo mutado (citosina, en azul) está presente en menor proporción en la madre que en el índice, lo que sugiere mosaicismo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Pedigrí de la familia estudiada junto con los electroferogramas de la región de FBN1que muestran la mutación c.2677+5G>C (flecha). El alelo mutado (citosina, en azul) está presente en menor proporción en la madre que en el índice, lo que sugiere mosaicismo. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Tras el consentimiento informado, se realiza estudio genético al índice mediante secuenciación masiva de 30 genes relacionados con enfermedad vascular aórtica. Se identifica en heterocigosis una mutación no descrita previamente en el intrón 22 de FBN1 (c.2677+5G>C; NM_000138.4). Estudios in silico (SSF, MaxEnt, NNSplice, HFF) la consideran como posiblemente asociada a la enfermedad, al conllevar la pérdida del donador natural del splicing. Aunque esta variante no se ha descrito previamente en población general (dbSNP, Exome Variant Server), en la literatura se ha indicado otra mutación en la misma posición, pero con otro cambio nucleotídico, en un paciente con síndrome de Marfan, lo que sugiere la importancia de esta posición para el correcto procesamiento del ácido ribonucleico6. Además, nuestro paciente presenta otras 2 mutaciones de patogenicidad desconocida, en TGFBR1 (c.409G>A; p. Val137Ile) y en LMNA (c.1158-6C>T, NM_170707.3).
En el estudio familiar, la madre cumple criterios diagnósticos de síndrome de Marfan (afección aórtica y antecedente familiar) con una puntuación sistémica de 3 (escoliosis, miopía >3 dioptrías, pectus excavatum). Tiene 3 hermanas que no expresan fenotipo y ambos progenitores fallecidos hace años, a edad avanzada, aparentemente sin afección cardiovascular, aunque no estudiados.
En el estudio de cosegregación familiar se comienza con la madre, con estudio dirigido hacia FBN1 y TGFBR1 (este último, por considerarse gen relacionado con síndromes aórticos familiares), y resulta no portadora para TGFBR1 pero sí para FBN1, en mosaico (figura). La presencia de este mosaicismo somático permite interrumpir el estudio genético en cascada en sus hermanas, ya que este fenómeno implica que la mutación se produjo de novo en algunas células durante el desarrollo embrionario de la madre. Siguiendo las recomendaciones genéticas para los estudios de sospecha de mosaicismo, se realizaron estudios adicionales en otros tejidos (mucosa bucal) y también con una pareja de cebadores independientes, y se obtuvo un porcentaje similar para el alelo mutado, lo que sugiere que el suceso mutacional ocurrió en los primeros estadios de la embriogénesis.
Al revisar en la literatura médica los casos descritos hasta la fecha, el progenitor portador de la alteración en mosaico, independientemente del sexo, presentaba un fenotipo menos grave que el índice, o incluso ausente2–5. Sin embargo, en nuestra familia destaca la alta expresión fenotípica vascular en la madre, a pesar de ser mosaico. Si bien podría pensarse que esta discrepancia es consecuencia del tipo de alteración genética, los datos publicados no apoyan tal hipótesis ya que la menor expresión fenotípica de los progenitores mosaico está presente asociada a cualquier tipo de mutación (tanto de cambio de aminoácido como de proteína truncada)2–5. En consecuencia, consideramos necesario un estrecho seguimiento clínico de los pacientes aunque presenten la mutación en un bajo porcentaje de células.
Respecto a la variabilidad intrafamiliar, en nuestro caso podría explicarse por un posible efecto protector de la variante encontrada en TGFBR1 en el hijo, o por la diferencia de edad entre ambos en relación a la posible expresión tardía de esta enfermedad, ya que es conocido que la afección aórtica en el síndrome de Marfan es progresiva y que el embarazo, además, es un factor de riesgo añadido1.
En resumen, se presenta un ejemplo de mosaicismo somático en FBN1 que ilustra la importancia de considerar dicha posibilidad al realizar un adecuado consejo genético, permitiendo adoptar una estrategia en cascada más restringida de lo que se hubiera planteado inicialmente. Asimismo, el hallazgo del mosaicismo no siempre permite asegurar una evolución menos grave del síndrome de Marfan.
Los autores agradecen al paciente y sus familiares su colaboración para el desarrollo de este trabajo.