El síndrome de Marfan se produce principalmente por mutaciones del gen FBN1. El diagnóstico suele basarse en criterios clínicos, pero la forma de presentación fenotípica es muy diversa en los individuos afectados. La disección o la rotura aórtica son la causa de la muerte en más del 90% de los pacientes no tratados. La identificación precoz de los individuos en riesgo es importante, teniendo en cuenta la disponibilidad de tratamientos médicos y quirúrgicos que mejoran significativamente la esperanza de vida. Los estudios moleculares pueden proporcionar un diagnóstico etiológico en los pacientes con formas de presentación clínica atípicas o más leves y contribuyen al manejo preventivo de portadores y el consejo genético y la tranquilización de los individuos no afectados. En este artículo, mediante la descripción de una familia con síndrome de Marfan con una forma de presentación vascular atípica y agresiva, ponemos de relieve la utilidad de las pruebas de detección de la mutación de FBN1 en casos seleccionados.
Palabras clave
La disección o la rotura aórticas constituyen la principal causa de muerte en más del 90% de los pacientes con síndrome de Marfan (SMF) no tratados1, pero en alrededor del 18% de los pacientes el primer episodio aórtico (dilatación o disección) puede producirse en la aorta distal2.
El SMF tiene su causa principal en mutaciones del gen de la fibrilina-1 (FBN1), un gen grande situado en el cromosoma 15. En la última actualización de la Universal Marfan Database se registraban más de 600 mutaciones patógenas, y la mayoría de ellas se dan de forma específica en una sola familia3.
Hasta la fecha no se ha establecido ninguna correlación de genotipo-fenotipo. No obstante, el riesgo relativo de afección de órganos específicos muestra ciertas diferencias significativas entre los distintos tipos de mutaciones4.
La identificación temprana de los pacientes con riesgo de SMF es importante, dada la disponibilidad de tratamientos médicos y quirúrgicos que mejoran de manera significativa la esperanza de vida.
En este artículo describimos a una familia en la que varios miembros expresaban unas manifestaciones clínicas compatibles con el SMF, pero con unas características fenotípicas peculiares, y en la que el diagnóstico molecular permitió investigar a los familiares y establecer un diagnóstico definitivo en alguno de ellos.
MétodosDatos clínicosSe invitó a los pacientes y sus familiares a acudir a una consulta clínica con un cardiólogo, un reumatólogo y un oftalmólogo; el diagnóstico de SMF se realizó según los criterios de Ghent5.
El examen ecocardiográfico se llevó a cabo con un aparato de ecografía Vivid 3 (GE Healthcare) o iE33 (Phillips). Se midieron los diámetros aórticos según las recomendaciones realizadas para la cuantificación de las cámaras por la American Society of Echocardiography y la European Association of Echocardiography6. Se consideró que la raíz aórtica estaba dilatada si el diámetro máximo en los senos de Valsalva era superior a los límites normales para la edad y la superficie corporal7.
Se realizó una angiorresonancia magnética (angio-RM) de 3 T (Siemens Magnetom Trio) para la exploración corporal total en algunos de los pacientes. Se obtuvieron exploraciones en cortes transversales, longitudinales, coronales y sagitales oblicuos que incluían la aorta ascendente, el cayado aórtico y la aorta descendente. Se analizaron también reconstrucciones multiplanares.
Como criterio ocular sólo se tuvo en cuenta la ectopia del cristalino. No se realizaron exploraciones de resonancia magnética para buscar una posible ectasia de la duramadre.
Datos molecularesTras la obtención del pertinente consentimiento informado por escrito, se extrajo ADN de una muestra de sangre periférica y se amplificaron la totalidad de los 65 exones del gen FBN1. A continuación se realizó un primer examen de detección sistemática de mutaciones en toda la secuencia de codificación del gen FBN1, con el empleo de reacción en cadena de la polimerasa. Todos los resultados positivos se confirmaron con una repetición del análisis.
ResultadosDescripción de la familiaNuestro caso inicial (Figura 1, III:7), una mujer de 37 años de edad, fue remitido a nuestro centro tras presentar una disección aórtica de tipo B que se iniciaba 60 mm después del origen de la arteria subclavia izquierda y se extendía distalmente hasta las arterias iliacas; la aorta ascendente y el cayado aórtico estaban dentro de los límites normales y la aorta descendente torácica tenía un diámetro de 42 mm.

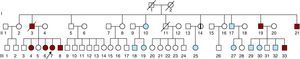
Figura 1. Árbol genealógico de la familia. Flecha: caso inicial. Símbolos rojos: individuos afectados que cumplen los criterios de Ghent. Símbolos con línea negra vertical: individuos afectados que no cumplen criterios de Ghent. Símbolos azules: exámenes clínicos y moleculares negativos. ?: individuo posiblemente afectado. Símbolos con una línea oblicua: individuos fallecidos.
Los antecedentes familiares obtenidos en ese momento indicaron que el padre (Figura 1, II:3) había fallecido de forma súbita a la edad de 48 años (tras sufrir una disección aórtica de tipo B) y que dos tíos (Figura 1, II:19, II:21) ya estaban en seguimiento para la detección de una posible afección aórtica. Al mayor de ellos, un varón de 53 años (Figura 1, II:19), se le implantó un injerto sintético Bentall a la edad de 50 años a causa de una dilatación progresiva de la raíz aórtica (60 mm). Un segundo tío de 48 años (Figura 1, II:21) presentó una enfermedad arterial generalizada. Se le amputó la pierna izquierda a causa de una arteriopatía oclusiva a la edad de 45 años y se le practicó exclusión de un aneurisma poplíteo de la pierna derecha a la edad de 46. También presentaba una dilatación leve de la aorta abdominal y un aneurisma de la raíz aórtica (34 y 46 mm de diámetro respectivamente).
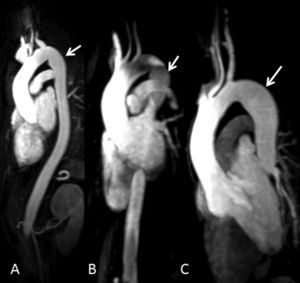
Por entonces se habían iniciado ya las pruebas de detección en la familia y se convocó a los hermanos de la paciente para un examen clínico. Tres de ellos (2 mujeres y 1 varón) fueron evaluados; una hermana de 43 años (Figura 1, III:4) tenía una aorta de un tamaño normal, pero con una ectasia postístmica peculiar según lo observado en la RM (Figura 2A); una hermana de 39 años (Figura 1, III:6) presentaba una dilatación leve de la raíz aórtica (40 mm), una aorta descendente de tamaño normal y la misma ectasia postístmica peculiar (Figura 2B); el hermano varón más joven (Figura 1, III:8), un paciente de 36 años, presentaba una dilatación de la raíz aórtica (47 mm) y una ectasia aórtica discreta (31 mm) de la aorta descendente inmediatamente después del istmo (Figura 2C). Cuatro días después de realizar nuestro examen clínico y de diagnóstico por la imagen, este paciente fue ingresado por una disección aórtica aguda de tipo B, que se iniciaba inmediatamente después del origen de la arteria subclavia; la tomografía computarizada puso de manifiesto una dilatación aórtica (44 mm) a la altura de la disección.

Figura 2. Reconstrucción tridimensional de la angiorresonancia magnética de los pacientes III:4 (A), III:6 (B) y III:8 (C) de la figura 1. Se aprecia un leve estrechamiento peculiar a nivel del istmo y una ectasia aórtica postístmica (flechas).
Un primo hermano de la paciente inicial (Figura 1, III:33), de 19 años de edad, sufrió una disección aguda de tipo A (a la edad de 19 años) que se extendía por toda la aorta; la aorta ascendente mostraba una intensa dilatación (82 mm) y el paciente fue remitido a una intervención quirúrgica de urgencia para la sustitución de la raíz de la aorta y la válvula aórtica.
En una tía de la paciente inicial, de 61 años de edad (Figura 1, II:14), se observó dilatación de la raíz aórtica (43 mm).
Las características clínicas de los familiares afectados se indican en la Tabla 1. Los demás familiares examinados (12 individuos) no presentaban signos clínicos ni moleculares de SMF.
Tabla 1. Manifestaciones clínicas de los miembros de la familia afectados
| Paciente | Sexo | Edad | Sistema cardiovascular | Sistema esquéletico | Sistema ocular (ectopia de cristalino) | Pulmón (neumotórax espontáneo) | Piel | ||||||||||
| DilA | DisA | PVM | PCar | PExc | LB/T | Escol | MyP | PPlan | PAcet | Hart | CaFc | ||||||
| II:14 | M | 61 | + | − | − | − | − | 1,03 | − | − | − | ND | − | − | ND | − | − |
| II:19 | V | 53 | +/Cir | − | +/Cir | ND | ND | 1,08 | + | + | + | + | − | − | − | − | − |
| II:21 | V | 48 | + | − | − | ND | ND | 1,03 | − | + | − | + | − | + | − | + | + |
| III:4 | M | 43 | − | − | + | − | + | 1,04 | + | + | + | + | − | + | − | – | + |
| III:6 | M | 39 | + | − | − | − | + | 1,02 | +/Cir | + | + | + | + | − | − | + | + |
| III:7 | M | 37 | − | + | + | − | + | 1,04 | + | + | + | + | + | + | − | − | + |
| III:8 | V | 36 | + | + | + | + | − | 1,06 | + | + | + | + | + | − | − | − | + |
| III:33 | V | 19 | + | +/Cir | − | − | − | 1,08 | − | + | − | + | + | + | − | − | + |
−: ausente; +: presente; CaFc: características faciales; Cir: cirugía; DilA: dilatación aórtica; DisA: disección aórtica; Escol: escoliosis; Hart: hipomotilidad articular; LB/T: proporción de longitud de brazo respecto a altura; M: mujer; MyP: signos de muñeca y pulgar; ND: no disponible; PAcet: protrusión acetabular; PCar: pectus carinatum; PExc: pectus excavatum; PPlan: pies planos; PVM: prolapso de válvula mitral; V: varón.
Se identificó una nueva mutación patógena del gen FBN1 en todos los miembros afectados de esta familia. La hallada era una mutación carente de sentido, con sustitución C1995G en el exón 16 del gen FBN1, que comportaba la formación de un codón de detención en la posición 665 de tirosina (Tyr665X).
DiscusiónEsta familia fue conocida por nuestro centro a causa de dos casos de disección aórtica de tipo B (Figura 1, III:7, III:8). Los diámetros aórticos eran menores que los esperados en esas complicaciones y la aorta descendente tenía una anatomía peculiar, para la que no se había descrito anteriormente una asociación con el SMF (Figura 2). Además, en otro de los miembros de la familia hubo un diagnóstico inicial de arteriopatía periférica (aneurisma poplíteo) y se le diagnosticó más tarde una dilatación de la aorta abdominal y ascendente. Se sospechó un síndrome arterial generalizado de carácter familiar y se realizó una búsqueda de mutaciones en los genes FBN1 y TGFβR. La identificación molecular de una mutación en el gen FBN1 fue determinante para establecer un diagnóstico definitivo de SMF en esta familia.
Las mutaciones del gen FBN1 se identifican en alrededor del 90% de los pacientes con SMF que cumplen los criterios de Ghent8, pero se ha observado también a pacientes con los fenotipos clásicos del SMF que son portadores de mutaciones en los genes TGFβR1 y TGFβR2 (SMF tipo 2), las mismas que causan el síndrome de Loyes-Dietz (SLD). Es característico que estos pacientes presenten una tortuosidad arterial y aneurismas de manera generalizada, así como que sufran una disección aórtica a una edad temprana y con diámetros aórticos menores9, pero se ha observado un amplio solapamiento clínico entre el SMF, el SMF tipo 2 y el SLD10.
En esta familia, observamos una nueva mutación del FBN1 que producía una truncación temprana de la secuencia proteica y se asociaba a un fenotipo cardiovascular especialmente agresivo en la mayor parte de los individuos afectados. La mutación terminadora (nonsense mutation) hallada estaba situada en el exón que codifica el segundo de siete módulos del tipo de la proteína transportadora de TGFβb1 de la fibrilina 1, dominio este que sólo está presente en FBN1 y los LTBP. Estos dominios globulares no tienen ninguna función específica conocida, aunque parecen intervenir en interacciones específicas entre proteínas11.
Actualmente se piensa que el aumento de la actividad de TGFβb es un importante factor contribuyente en la patogenia tanto del SMF como del SLD9,12.
En el futuro, es posible que un mejor conocimiento del fundamento genético y las vías moleculares involucradas puedan modificar nuestra forma de abordar la enfermedad aórtica en el SMF. Mientras tanto, los criterios actualmente utilizados para la intervención quirúrgica sobre la aorta en los pacientes con SMF con un diámetro de la aorta ascendente ≥ 5 cm y un diámetro de la aorta descendente ≥ 6 cm13. Estos valores están muy lejos de las dimensiones aórticas que se daban en nuestro caso inicial y en su hermano, pero ello no impidió que sufrieran disección aórtica de tipo B. La cuestión de si debe intervenirse o no a las otras dos hermanas que presentan una morfología aórtica similar y, en su caso, cuándo hacerlo es difícil, y en este momento se encuentran en estricta vigilancia clínica y con exploraciones de imagen, con objeto de detectar cualquier posible progresión de las dimensiones aórticas. En resumen, en esta familia, la detección de una mutación del gen FBN1 reveló el carácter genético de las manifestaciones fenotípicas descritas, lo que permitió el diagnóstico de pacientes con formas de presentación clínica más leves y contribuyó a facilitar un diagnóstico precoz, un consejo genético y el tratamiento preventivo de los portadores o la tranquilización de los individuos no afectados. El diagnóstico diferencial entre el SMF clásico, el SMF2 o un subtipo de SLD no habría sido posible con medios exclusivamente clínicos.
FinanciaciónEste estudio contó con la financiación de una subvención del Departamento de Investigación del Hospital S. João de Oporto.
Conflicto de interesesNinguno.
Recibido 2 Enero 2010
Aceptado 21 Febrero 2010
Autor para correspondencia: Hospital de S. João. Alameda Prof. Hernâni Monteiro, 4202-451 Oporto, Portugal. ana.lebreiro@gmail.com