Palabras clave
INTRODUCCIÓN
Durante más de mil años, el concepto de circulación pulmonar se basó en las enseñanzas de Galeno,que consideraba que la sangre se generaba en el hígado y, a continuación, el ventrículo derecho (VD)la repartía por los tejidos y órganos donde se consumía. Según Galeno, la sangre «se filtraba» haciael ventrículo izquierdo (VI) directamente desde elVD a través de poros invisibles en el tabique interventricular. Aunque hoy en día pueda resultar evidente que esta teoría es imposible, Galeno veía elmovimiento sanguíneo como flujo y reflujo de bajovolumen1.
En el siglo XIII, Ibn al-Nafis de Siria rechazó laexplicación de Galeno y especuló con que la sangredel VD llegaría al VI a través de los pulmones.Aunque merece ser reconocido por proponer la primera descripción acertada de la circulación pulmonar, sus obras se perdieron y se mantuvieron enun largo olvido hasta hace poco, y no parece probable que influyeran en la comprensión de la fisiología circulatoria en Occidente2.
La primera descripción detallada del VD y de lacirculación pulmonar que recibió una atención significativa en Occidente surgió casi a principios delsiglo xvi en una disertación religiosa del españolMiguel Servet. Debido a su obra, Servet fue posteriormente condenado a morir en la hoguera,aunque seguramente más por el carácter herético desu contenido religioso que por sus opiniones acercade la fisiología circulatoria. En ella, Servet escribió que:
«[El espíritu vital] tiene su propio origen en elventrículo izquierdo del corazón, y los pulmonestienen un papel importante en su desarrollo. Setrata de un espíritu enrarecido, producido por lafuerza del calor, de color amarillo rojizo (flavo) yde potencia igual a la del fuego. De manera que esuna especie de vapor de sangre muy pura que contiene en sí mismo las sustancias del agua, aire yfuego. Se genera en los pulmones a partir de unamezcla de aire inspirado con la sangre elaborada yligera que el ventrículo derecho del corazón comunica con el izquierdo. Sin embargo, esta comunicación no se realiza a través de la pared central del corazón, como comúnmente se cree, sino que, a travésde un sistema muy ingenioso, la sangre fluye durante un largo recorrido a través de los pulmones.Elaborada por los pulmones, adquiere el tono amarillo rojizo y se vierte desde la arteria pulmonarhasta la vena pulmonar»3.
Este modelo, estrictamente basado en observaciones estructurales más que en mediciones experimentales, se aparta radicalmente de Galeno, pero aligual que hiciera Galeno anteriormente, Servetasumió que la sangre se producía y se consumíacontinuamente, en lugar de recircular3.
Cincuenta años más tarde, William Harvey desarrolló el primer modelo de circulación basadoen experimentos. Pese a no ser la primera personaque describiera la circulación pulmonar, Harveyse considera el padre de la fisiología moderna, yaque fue el primero que realizó mediciones y cálculos detallados1 que le permitieron deducir laexistencia de la recirculación de la sangre y demostrar el flujo sanguíneo pulmonar de formaexperimental4.
Durante los 400 años siguientes se seguiría debatiendo acerca de la importancia del VD y, ya entradoel siglo XX, algunos investigadores seguían opinandoque la única función del VD era proporcionar capacidad a la circulación pulmonar5,6. Tales investigaciones tempranas dieron lugar en gran medida a lacreencia de que la insuficiencia cardiaca derechaconstituía un problema limitado principalmente a lahipertensión pulmonar idiopática y a la cardiopatíacongénita, una causa de muerte común. Sin embargo, ahora sabemos que la hipertensión pulmonar(HP) y la insuficiencia cardiaca derecha, lejos de serinfrecuentes, complican muchos otros procesos patológicos: la insuficiencia del VD es uno de los factoresde predicción de mortalidad más fiables en la insuficiencia cardiaca izquierda7. La insuficiencia cardiacaderecha es la causa de la muerte de la mayoría de los 50.000 casos mortales de embolia pulmonar quetienen lugar en Estados Unidos cada año8 y, segúnciertas estimaciones, 2-6 personas/1.000 con enfermedad pulmonar crónica padecerán insuficienciacardiaca derecha, con decenas de miles de casosnuevos al año9.
Este trabajo analiza la forma en que la interacción entre la circulación pulmonar y el VD contribuye al impacto de éstos en la salud y la enfermedad.
DESARROLLO DE LA CIRCULACIÓN PULMONAR Y NEONATAL Y DEL VENTRÍCULO DERECHO
En la tercera semana de la gestación humana, ladifusión pasiva de oxígeno en el embrión en desarrollo resulta insuficiente para soportar el metabolismo, la sangre se ha formado y el tubo cardiacoprimitivo ha comenzado a latir; y a finales de lacuarta semana comienza la circulación activa. Entrela tercera y la quinta semana de gestación surgendistintos componentes de la circulación pulmonar ysistémica a partir de los pliegues y giros del tubocardiaco primitivo, controlados por una complejared de señales que incluye las vías del ácido retinoico y la neuregulina. Poco después, el VD y lacirculación pulmonar empiezan a separarse del VI yla circulación sistémica por la formación del tabique interventricular a partir del cojín endocárdico, y se desarrollan las válvulas. En el nacimiento,suele haber concluido la septación completa del tabique interauricular y sólo el foramen oval permanece como comunicación potencial entre la aurículaderecha y la izquierda10-12.
En el embrión y el feto, el VD es la cámara dominante, ya que moviliza el 60% del gasto cardiacototal. Dado que el embrión recibe oxígeno y nutrientes de la placenta, sólo entre el 15 y el 25% delgasto cardiaco total entra en los pulmones. El restodel gasto cardiaco derecho pasa a la circulación sistémica, tanto a través del foramen oval hacia la aurícula izquierda como a través del conducto arteriosodesde la arteria pulmonar a la aorta. Entre el 40 y el60% del flujo aórtico descendente entra en la placenta a través de la arteria umbilical y a continuaciónvuelve por la vena umbilical al hígado o a través delconducto venoso a la vena cava inferior13,14.
En el nacimiento, la resistencia vascular pulmonar desciende rápidamente tras la expansión yoxigenación de los pulmones, y el gasto cardiacoventricular derecho empieza a fluir principalmentea través de la arteria pulmonar hacia los pulmones.En ese momento, el aumento de la presión auricularizquierda provoca el cierre de la «válvula de lengüeta» unidireccional que es el foramen oval15. Al nacer, las presiones del VD siguen sobrepasando laspresiones sistémicas, pero éstas comienzan a disminuir a las pocas horas o días16. Poco después, el ductus arterioso, controlado por la prostaglandina,empieza a cerrarse14, el VI se hipertrofia a medidaque se hace cargo de la circulación sistémica y elVD se atrofia. A la edad de 3 semanas, generalmente la presión pulmonar ha disminuido por debajo de la presión sistémica y en la edad adulta el VD normal es incapaz de generar más de40-60 mmHg17.
CIRCULACIÓN PULMONAR ADULTA
La arteria pulmonar consiste en un vaso fino y elástico que se ramifica para abastecer a las arterias lobulares pulmonares, las arteriolas pulmonares y los capilares alveolares. La sangre sale de los capilares alveolares a través de las vénulas pulmonaresy vuelve a la aurícula izquierda a través de un sistema de vénulas y ramificaciones pulmonares conestructura similar a la del árbol arterial pulmonar18-20. Dado que el intercambio de gases se produce en membranas finas y altamente permeables,la presión pulmonar debe ser baja para evitar eledema pulmonar por fuerzas de Starling elevadas21.
A diferencia de la circulación sistémica, en la quela capa media circunferencial de células de músculoliso de las arteriolas regula claramente la resistencia, las arteriolas pulmonares de menos de 70 μmde diámetro parecen tener como mucho capas incompletas de músculo liso en la media, lo cual llevóen el pasado a suponer que la regulación del flujosanguíneo se limitaba a vasos de más de 100 μm dediámetro. Sin embargo, otros estudios han mostrado que dicha regulación también se produce enlos microvasos pulmonares de entre 30 y 200 μm22. La regulación está controlada por un mecanismopoco conocido, sensible al oxígeno, que puede ser mediado por canales de potasio dependientes delcalcio o del voltaje, radicales de oxígeno u otrosmecanismos23, así como del óxido nítrico, las prostaglandinas, la endotelina y las catecolaminas24-26.
Generalmente se aplica una serie de conceptospara describir la resistencia al flujo en la circulaciónpulmonar y la «poscarga» consiguiente que se produce en el VD. La descripción más completa la proporciona la impedancia de entrada pulmonar. Sinembargo, la caracterización completa de la impedancia de entrada es muy compleja desde un puntode vista técnico, ya que requiere analizar el dominiode la frecuencia de la presión y del flujo midiéndolos simultáneamente27,28. No es fácil relacionar los factores derivados del dominio de la frecuenciacon conceptos clínicos comunes, por lo que generalmente se prefieren modelos simplificados. El denominado modelo de Windkessel29 abarca tres de loscomponentes principales de la impedancia de entrada: resistencia vascular pulmonar (RVP), distensibilidad arterial pulmonar y un componente dinámico denominado inductancia.
La RVP se define como la caída media de presiónde la arteria pulmonar principal a la aurícula izquierda dividida por el gasto cardiaco medio, expresada en dyn/s/cm5. Para facilitar el cálculo,suelen utilizarse unidades Wood, definidas como lapresión arterial pulmonar media menos la presiónde enclavamiento capilar pulmonar media enmmHg, dividida por el gasto cardiaco en l/min.Una unidad Wood equivale a 80 dyn/s/cm5. LaRVP está causada principalmente por la resistenciade los vasos pequeños, aunque la compresión extrínseca o la obstrucción mecánica de arterias másgrandes (p. ej., por embolia) también puede alterarla RVP. La distensibilidad arterial pulmonar se refiere a las propiedades elásticas del sistema y se define como la proporción entre un cambio en el volumen y un cambio en la presión; la distensibilidadarterial pulmonar amortigua el flujo durante la expulsión del VD y reduce la presión del pulso de laarteria pulmonar. La inductancia describe la respuesta dinámica a cambios en el flujo debidos a lamasa y la inercia de la sangre.
En los llamados modelos de parámetros agrupados (entre ellos el modelo de Windkessel), la resistencia se modela por medio de una o más resistencias eléctricas, la distensibilidad se modela pormedio de un condensador y la inductancia pormedio de un inductor o como resistencia en modelos más simplificados. Es posible realizar distintascombinaciones de estos elementos, pero generalmente se utilizan tres o cuatro modelos elementalesen las investigaciones fisiológicas. Los modelos de Windkessel proporcionan estimaciones bastantemejores del comportamiento del sistema que la resistencia vascular pulmonar por sí sola.
Hay otros términos que se utilizan normalmenteen los estudios fisiológicos, aunque la incoherenciaexistente en su definición en la literatura puedellevar a confusiones. La elastancia arterial efectivase define como la proporción entre la presión sistólica final con el volumen de latido, aunque habitualmente el término «elastancia» es simplemente lomismo que la distensibilidad. Esta sencilla mediciónagrupa componentes estáticos y dinámicos de impedancia y funciona bastante bien en estudiosexperimentales30,31. Aunque su uso ha sido más validado en el modelado de la circulación sistémica,también ha sido aplicado con éxito a la circulaciónpulmonar32.
HIPERTENSIÓN PULMONAR
La tabla 133,34 muestra las presiones y resistenciastípicas de las circulaciones pulmonar y sistémica.En condiciones normales, la RVP es 1/20 de la resistencia vascular sistémica, y la presión arterialpulmonar media no puede ser mucho más alta quela presión venosa central. Dado que un gradiente depresión de 5 mmHg a través de la circulación pulmonar basta para mantener un gasto cardiaconormal en condiciones normales de RVP y presiones de llenado del VI normales, normalmente esnecesaria la función contráctil mínima del VD paramantener el gasto cardiaco, lo que permite cirugíasde reparación de cardiopatías congénitas del tipoFontan, en las que el VD queda completamente excluido de la circulación pulmonar35.

En contraste con la atención a la impedancia vascular pulmonar prestada por los fisiólogos experimentales, los médicos se centran principalmente en la presión arterial pulmonar como concepto operativo fundamental, definiendo la hipertensión pulmonar (HP) como la presión arterial pulmonarmedia > 25 mmHg, o una presión máxima> 35 mmHg. Sin embargo, la presión arterial pulmonar aumenta con la edad, el intervalo de normalidad es amplio36 y la presión arterial pulmonar esuna función de la resistencia vascular pulmonar, elgasto cardiaco y la presión a la salida de las venas pulmonares. Por lo tanto, centrarse únicamente enla presión de la arteria pulmonar oculta la etiologíay las posibles opciones terapéuticas para la HP.
En el pasado, la HP se dividía conceptualmenteen aguda y crónica, y en primaria y secundaria.Dado que estos términos no proporcionaban información sobre su etiología o la posible terapia, laOrganización Mundial de la Salud desarrolló unnuevo sistema de clasificación que se resume en latabla 237,38.

El grupo I de la HP (conocido generalmentecomo hipertensión arterial pulmonar) se definecomo la HP que surge de anomalías primarias en laanatomía o la función de la vasculatura pulmonar.Ello incluye hipertensión arterial pulmonar idiopática (anteriormente conocida como «hipertensiónpulmonar primaria», término actualmente endesuso por los especialistas en HP pero que aún seutiliza). El grupo I de HP se debe generalmente aanomalías en la pared vascular de las arteriolas pulmonares, aunque también incluye la enfermedadvenooclusiva pulmonar. Los cambios histológicosconcretos subyacentes (analizados en detalle en otrolugar24,39) varían en cierta medida dependiendo dela etiología, pero en la mayor parte de los casos parece producirse una obstrucción mecánica al flujo yuna reactividad reducida a los vasodilatadores, loque se conoce como HP «fija» (si bien se trata de untérmino equivocado, ya que el proceso puede versemodificado por diversas terapias). A pesar de la escasa presencia, comparativamente, de la HP idiopática (con miles de nuevos casos anuales en todo elmundo), esta entidad recibe de los especialistas enHP un grado de atención desproporcionado.
El grupo II de HP (conocido generalmente comohipertensión venosa pulmonar) se define como HPdebida a la transmisión retrógrada de presionesanormalmente elevadas a las venas pulmonares pordiferentes causas. El grupo II de HP suele derivarde anomalías de la anatomía o la función del ladoizquierdo del corazón —por ejemplo, disfunciónsistólica o diastólica del VI o enfermedad de la válvula mitral y aórtica— y se trata fundamentalmentede un proceso pasivo con respecto a la vasculatura pulmonar. El grupo II de la HP puede «revertirse»teóricamente mediante la corrección del procesopatológico subyacente que condujo a una presiónelevada en la vena pulmonar. Sin embargo, en muchas situaciones (p. ej., estenosis mitral), los aumentos en la resistencia de la arteriola pulmonarpueden desarrollarse con el tiempo y hacerse irreversibles, supuestamente debido a mecanismos similares a los de ciertos tipos del grupo I de la HP.Aunque el grupo II de la HP es muy común (dehecho, algunos autores afirman que la causa másfrecuente de insuficiencia cardiaca derecha es la insuficiencia cardiaca izquierda), no se sabe con certeza en qué medida contribuye realmente la insuficiencia cardiaca derecha en el grupo II a lamortalidad y en qué medida constituye simplementeun indicador de insuficiencia cardiaca izquierdamás avanzada.
Los grupos III y IV de la HP se deben a alteraciones en las arteriolas pulmonares precapilares,pero en el grupo III dichas alteraciones derivan deenfermedades pulmonares o hipoxemia, y hastacierto punto se puede considerar que es una respuesta fisiológica normal a estímulos externos. Lavasoconstricción pulmonar hipóxica, mecanismoque normalmente adapta la perfusión pulmonar ala ventilación pulmonar, puede volverse patológicacuando demasiados segmentos del pulmón sevuelven hipóxicos y la RVP aumenta demasiado.La HP debida a procesos del grupo III puede revertirse por vasodilatadores como bloqueadores de canales de calcio, vasodilatadores pulmonares directos como el nitroprusiato y agentes inhaladoscomo el óxido nítrico, pero con el tiempo puedendesarrollarse anomalías del sistema vascular pulmonar y volverse prácticamente permanentes como en el grupo I de la HP. Además, la reversión de lavasoconstricción pulmonar hipóxica a través de medios extrínsecos puede empeorar la hipoxemia porun incremento del desajuste en la relación ventilación/perfusión. El grupo III de la HP probablemente representa más casos de HP que todos losdemás grupos juntos, aunque la gravedad del grupoIII de la HP tiende a ser algo menor que en losgrupos I o IV9.
El grupo IV de la HP se debe a la obstrucciónmecánica de arterias o arteriolas pulmonares derivadas de embolia pulmonar (crónica o aguda) yembolia tumoral. No hay estadísticas globales disponibles para este grupo, pero en Estados Unidospuede haber más de 600.000 casos de embolia pulmonar, que causan más de 60.000 muertes cadaaño8. El grupo V de la HP es un cajón de sastre queabarca procesos totalmente desconocidos o que noencajan en las categorías anteriores.
ANATOMÍA DEL VENTRÍCULO DERECHO ADULTO
La anatomía del VD ha sido revisada en detallepor Ho et al40. Conceptualmente, el VD puede dividirse en un tracto de entrada (que comienza en elanillo tricúspide), una región apical y un tracto desalida del VD (que acaba en la válvula pulmonar).La pared libre del VD constituye el borde anteriordel VD y consiste en media circunferencia relativamente fina de músculo, situada anterior al VI y altabique interventricular. El VD suele tener ungrosor < 1-3 mm, en comparación con los 10 mmde grosor de la pared libre ventricular izquierda, ycomprende casi 1/6 de la masa total del corazón17. Funcionalmente, el tabique interventricular constituye la otra mitad del VD. Los fascículos musculares espirales forman una estructura contigua enforma de banda que une funcionalmente el VD y elVI, lo que da como resultado la transmisión defuerza contráctil directamente del VI al VD17,41. Los fascículos transversales del eje corto del corazónvan del vértice a la base, con formas que varíandesde contornos prácticamente triangulares en elvértice a un aspecto de semiluna en la base. Estaforma compleja refleja la dificultad de evaluar el tamaño y la función del VD basándose en técnicas deimagen bidimensionales40, y también ilustra losgrandes cambios en el tamaño y la forma del VDque se producen con diversas condiciones de carga.
CIRCULACIÓN CORONARIA DEL VENTRÍCULO DERECHO
En la especie humana, el VD se perfunde en granmedida a través de la arteria coronaria derecha. Enel VI, la perfusión miocárdica se produce sobretodo en diástole cuando la presión del tejido intramiocárdico desciende por debajo de la presión de laraíz aórtica. En condiciones de carga normales, lapresión del tejido intramiocárdico del VD permanece por debajo de la presión de la presión de laraíz aórtica a lo largo del ciclo cardiaco, permitiendo un flujo coronario continuo, pero en caso desobrecarga aguda de presión del VD, el patrón deperfusión coronaria del VD empieza a acercarse aldel VI42.
CONTRACCIÓN NORMAL DEL VENTRÍCULO DERECHO
En el VI, el desarrollo de la presión ventricular yla expulsión de sangre se deben a la contracciónconcéntrica de la pared libre y el tabique del VI,junto con un movimiento de giro o «retorcimiento»del corazón. Por el contrario, la expulsión de sangrepor el VD se produce con una contracción secuencial que comienza en el tracto de entrada y se mueveen ondas hacia el tracto de salida17,43. La expulsión normal del VD es obra tanto de una reducción en lasuperficie de la pared libre del VD como de una reducción en la distancia del septo a la pared libre delVD44,45.
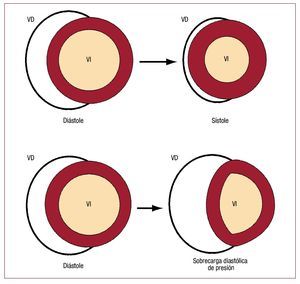
La figura 146 esquematiza cómo el VD y el VI expulsan sangre (omitiendo cualquier movimiento degiro o ventricular). Dado que el área de la superficiede un cilindro es proporcional a su radio y el volumen es proporcional al cuadrado del radio, lafracción de expulsión en el VI es prácticamente proporcional al cuadrado del cambio en el área de la superficie endocárdica. Por el contrario, dada lamayor proporción entre superficie de área y volumen del VD, se produce una mayor fracción deexpulsión por un cambio menor en el área de superficie que el que se necesitaría en el VI. La disposición en fuelle del VD no sólo permite grandes cambios en el volumen del VD con pequeños cambiosen el área de superficie de la pared libre del VD,sino que también ayuda a amortiguar cambios respiratorios en la salida del VD47 sin necesidad de alterar la función contráctil respiración a respiración.

Fig. 1. Ilustración de los cambios de forma en el corazón durante la contracción.La sección circular del VI se contrae poruna reducción uniforme de la superficieendocárdica, manteniendo una relacióncasi constante entre el volumen y el áreasuperficial. El VD con forma semilunar seaplana durante la sístole, lo que lleva a ungran cambio de volumen con un cambiomínimo en el área de la pared libre del VD.Durante la sobrecarga grave de presión,el tabique interventricular se desplaza eincrementa el volumen diastólico del VDcon un pequeño aumento en el área superficial de la pared libre del VD. Sin unincremento en el área, el VD no puedeaumentar su contractilidad por el mecanismo de Frank-Starling. Al mismo tiempo, se produce una reducción en el área yel volumen sistólico final del VI que derivaen deficiencias en la función de bombeodel VI. (Tomada de Greyson CR. Crit CareMed. 2008;36:S57-65. Copyright 2008,con permiso de Lippincott, Williams & Wilkins46). VD: ventrículo derecho; VI: ventrículo izquierdo.
Mientras que la configuración en serie de la circulación pulmonar y sistémica requiere que el volumen de latido medio del VD y del VI sea el mismo(en ausencia de cortocircuitos intracardiacos), elvolumen diastólico final del VD suele ser algomayor que el volumen diastólico final del VI, mientras que la fracción de expulsión es menor. A medida que la poscarga aumenta, el volumen diastólico final del VD se eleva, mientras que la fracciónde eyección se reduce48.
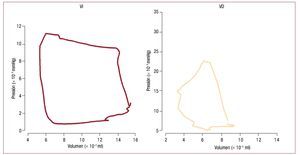
La dinámica de la contracción del corazón sueleanalizarse en el contexto de curvas de relación entrepresión y volumen, como muestra la figura 249. La forma cuadrada del bucle de la relación entre presión y volumen del VI sugiere la definición de lasfases de contracción y relajación isovolúmétrica(volumen constante) y simplifica la identificaciónde la telesístole y el cierre de la válvula aórtica, queocurren cerca del punto de inflexión de la fase de eyección. Suga y Sagawa desarrollaron la metodología para describir la función ventricular izquierdaen términos de una «elastancia variable en eltiempo», en la que la elastancia es igual a la pendiente de la relación presión-volumen en momentosespecíficos («isocrónica») del ciclo cardiaco50. Experimentalmente, se ha descubierto que la pendiente máxima de la relación presión-volumen delVI, llamada Emáx, suele producirse en la telesístole yestá directamente relacionada con el estado contráctil del VI49. En la mayoría de los casos equivaleesencialmente a la elastancia telesistólica (Esf). Por el contrario, la expulsión de sangre a través de laválvula pulmonar puede continuar incluso cuandola presión del VD decae debido a la inercia de lasangre en el circuito pulmonar de baja impedanciade entrada. Esta eyección tardía o «periodopasivo»51 hace que la identificación de la «telesístole» sea problemática en el VD y contribuye a unaforma más triangular de la curva presión-volumendel VD. El resultado es que las curvas presión-volumen son más difíciles de interpretar en el VD queen el VI49,52 y que la relación volumen-presión telesistólica no es necesariamente el método de elecciónpara evaluar la función contráctil del VD53.

Fig. 2.Comparación de las curvas de presión-volumen obtenidas en humanos con catéteres con micromanómetros y ventriculografía del VI (izquierda) y VD(derecha). Los bucles de presión-volumen en el VI son casi cuadrados, lo que simplifica la identificación de las fases de contracción y relajación isovolumétricas. Por el contrario, el bucle del VD es más triangular, con telesístole pobremente definida. (Tomada de Redington AN, et al. Br Heart J. 1998;59:23-30.Copyright 1988, con permiso de BMJ Publishing Group Ltd.49). VD: ventrículo derecho; VI: ventrículo izquierdo.
ACOPLAMIENTO VENTRICULOVASCULAR
En cualquier sistema mecánico o eléctrico, latransmisión de energía de una parte del sistema a otra se maximiza cuando la impedancia de salida dela parte productora de energía y la impedancia deentrada de las partes receptoras de energía del sistema son iguales. Como se ha descrito anteriormente, la elastancia está relacionada con la impedancia, de manera que la máxima transferencia deenergía del ventrículo al sistema vascular se consigue si la elastancia ventricular (Emáx) y la vascular (Ea) son iguales54. Sin embargo, en el corazón palpitante, donde las propiedades mecánicas cambiancon el tiempo, los estudios teóricos y experimentaleshan demostrado55 que la máxima eficiencia de producción de trabajo (es decir, la relación entre trabajo sistólico y consumo de oxígeno) se alcanzacuando la proporción entre Emáx y Ea se acerca a 2. En condiciones normales, se ha descubierto experimentalmente que el acoplamiento ventriculovascular de la circulación sistémica prácticamente alcanza la máxima eficiencia. La telesístole del VD noestá tan claramente definida y, en consecuencia, laEmáx es más difícil de definir. Ahora bien, varios investigadores han descubierto que el acoplamientopulmonar vascular del VD puede analizarse de unmodo similar y que, si la telesístole se define demodo práctico, el acoplamiento también es prácticamente óptimo en condiciones normales (es decir,la relación entre la Emáx del VD y la Ea de elastanciade la arteria pulmonar se acerca a 2)56,57.
CONTRIBUCIÓN DEL VENTRÍCULO DERECHO AL GASTO CARDIACO
En 1943, Starr et al5 lesionaron el VD en perroscon el tórax abierto con un «soldador al rojo vivo»y hallaron pocas alteraciones en la circulación sistémica o la presión venosa. Conjeturaron que lapared libre del VD servía para proporcionar capacidad a la circulación pulmonar. Otros investigadores, por su parte, llegaron más tarde a conclusiones similares6, por lo que comenzó a decaer elinterés en el VD.
Sin embargo, el desarrollo de la presión del VDes una combinación de interacciones entre la paredlibre del VD, el tabique interventricular y la paredlibre del VI58,59, y la importancia de la función contráctil de la pared libre del VD depende en gran medida de la resistencia vascular pulmonar y la presión del VD. Por ejemplo, mientras que la oclusiónde la arteria coronaria derecha (ACD) y la disfunción contráctil de la pared libre del VD puedentener poco impacto en el desarrollo de presión porel VD o la hemodinámica sistémica en condicionesnormales, la isquemia del VD deriva en hipotensiónsistémica cuando aumenta la resistencia vascularpulmonar60. La prueba experimental de la importancia de la función contráctil del VD obtuvo unmayor reconocimiento tras las series de casos de Cohn de infartos aislados de VD con inestabilidadhemodinámica, y la evidencia posterior de que ladisfunción contráctil del VD en el contexto de uninfarto de miocardio (IM) derivaba en un aumentosustancial de la morbilidad y la mortalidad61,62.
RESPUESTA DEL VENTRÍCULO DERECHO A UN INCREMENTO AGUDO DE LA PRESIÓN
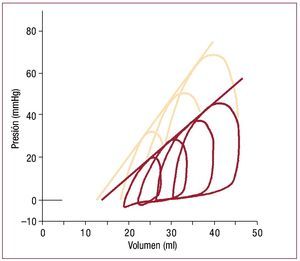
Durante más de un siglo se ha estudiado cómoresponde el corazón a la sobrecarga de presión, empezando por los esfuerzos de Otto Frank para sistematizar el estudio de la función ventricular63. Suga y Sagawa ampliaron y desarrollaron estos conceptos durante las décadas de los sesenta y los setenta, hasta tal punto que la relación entre volumeny presión es hoy objeto de discusiones habituales enun contexto clínico64. La figura 3 muestra curvaspresión-volumen en un VD aislado a medida que sealtera la presión telesistólica. Cada bucle representaun solo ciclo cardiaco52. En términos generales,sobre una escala fisiológica, el volumen sistólicofinal es casi inversamente proporcional a la presiónsistólica final, mientras que el volumen-latido (y,por lo tanto, el trabajo por latido) aumenta conforme lo hace el volumen diastólico final.

Fig. 3. Ejemplo de curvas presión-volumen del VD obtenidas en el corazónlatiente aislado de un perro en condiciones basales (líneas rojas) y tras laestimulación inotrópica con adrenalina (líneas amarillas). Las series de curvas en cada caso se obtienen variando la resistencia de salida. Obsérveseque prácticamente puede trazarse una línea recta tangencial a cada bucleen una condición particular; aunque la telesístole es difícil de identificar,esta línea es prácticamente equivalente a la relación presión-volumen sistólica final obtenida en ventrículos izquierdos aislados, donde la pendientees un reflejo del estado contráctil subyacente. (Tomada de Maughan WL,et al. Circ Res. 1979;44:309-15. Copyright 1979, con permiso de WoltersKluwer Health52). VD: ventrículo derecho; VI: ventrículo izquierdo.
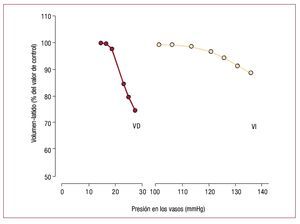
Sin embargo, en comparación con el VI, el VDadulto tiene una capacidad muy limitada para producir presiones elevadas. Dichos límites se ilustran en la figura 4, que muestra que el volumen-latidodisminuye mucho más deprisa en el VD que en elVI a medida que aumenta la presión media de expulsión65. La ley de Laplace ayuda a explicar lamenor capacidad del VD para contraerse frente auna carga. En primer lugar, la pared libre más finadel VD experimenta un mayor aumento en la tensión de la pared con incrementos en la presión delVD. En segundo lugar, el radio de curvatura delVD aumenta durante la contracción, en lugar dedisminuir como sucede en el VI, lo que significa quela reducción de la tensión en función de la formadurante la contracción debido al radio de curvaturadecreciente no se produce en el VD como en el VI66. También parece haber diferencias bioquímicas, yaque el miocardio del VD está más capacitado parala contracción rápida67, aunque todavía es inciertosi las diferencias en la composición de isoformas dela cadena pesada de la miosina explican este hecho,ya que las diferencias entre el VD y el VI en la expresión de las isoformas de la miosina están presentes en los roedores68, pero no en los perros69.

Fig. 4. Comparación del efecto de presiónde eyección en la fracción de eyección enel VD y el VI. Obsérvese que, para cualquierincremento en la presión de eyección,el descenso de la fracción de eyección es mucho mayor en el VD que en el VI. (Tomada de Heart Disease, Braunwald E,ed. by Wiederman HP, Saunders Company,1997, p. 1606, Copyright 1997, con permiso de Elsevier65). VD: ventrículo derecho; VI:ventrículo izquierdo.
Varios mecanismos pueden contribuir al incremento de la función contráctil en el contexto de unaumento de la demanda. Entre ellos figuran elefecto Anrep (autorregulación homeométrica), elmecanismo de Frank-Starling (conocido en ocasiones como autorregulación heterométrica) y elinotropismo inducido por catecolaminas.
El efecto Anrep es un incremento intrínseco de lafunción contráctil que se produce como respuesta aun incremento de la poscarga en ausencia de cambios regulatorios externos como la estimulación por catecolaminas. El efecto Anrep puede demostrarseen tiras aisladas de músculo69, pero su presencia invivo ha sido controvertida. Algunas pruebas indicanque el efecto Anrep es el mecanismo primario parala adaptación inicial a la sobrecarga de presión enel VD70,71, aunque puede ser más importante en elVD de neonatos que en el VD de adultos.
El mecanismo de Frank-Starling se ha considerado a menudo como el medio principal por el queel corazón se adapta a una mayor exigencia, perolas diferencias de forma entre el VD y el VI alteranel funcionamiento del mecanismo de Frank-Starling. Tanto en el VD como en el VI, un aumento en la presión sistólica final suele ir acompañado de un incremento tanto en el volumensistólico final como en el diastólico final. Sin embargo, en el VD en condiciones de carga normales,gran parte del aumento del volumen se debe a unaumento en la distancia entre el septo y la paredlibre del VD, con un incremento mucho menor enel área de superficie de la pared libre del VD.Dado que el incremento del área de pared libre delVD para un incremento determinado de la presiónvenosa central es pequeño, la implicación según elmecanismo de Frank-Starling es reducida. Por lotanto, el mecanismo de Frank-Starling desempeñaun papel menor en la adaptación del VD a un aumento en la poscarga con presiones del VD bajasque el que desempeña en el VI. Con una poscargaaumentada, a medida que el VD se vuelve más cilíndrico y se agotan otros mecanismos compensatorios, el mecanismo de Frank-Starling adquieremás importancia70,71. La estimulación simpática también incrementa la función contráctil en el VDal igual que en el VI52.
RESPUESTA DEL VENTRÍCULO DERECHO A UN INCREMENTO CRÓNICO DE LA PRESIÓN
Como ya se ha comentado, las presiones del VDsuelen ser mayores que las presiones sistémicas en elfeto y el neonato, pero disminuyen a los valores delos adultos durante los primeros días o las primerassemanas posteriores al nacimiento. Sin embargo,los VD de los neonatos pueden tolerar una HP persistente y los pacientes de cardiopatías congénitastoleran presiones suprasistémicas pulmonares durante años con escasas limitaciones (es el caso delsíndrome de Eisenmenger)72. Por el contrario, unavez que el VD se atrofia y la presión disminuye, losfuturos aumentos de presión (incluso los que se desarrollan en periodos prolongados) se toleran condificultad. La razón de esta diferencia sigue siendodesconocida.
La hipertrofia del VD puede desarrollarse en lassobrecargas crónicas de presión73, aunque se desconoce si ello ayuda a normalizar la tensión de paredo conduce a una disfunción contráctil74. Al mismo tiempo, la dilatación del VD puede conducir a unamayor contractilidad según la ley de Frank-Starling.
Se han detectado numerosas alteraciones bioquímicas en varios modelos de sobrecarga crónica depresión en el VD75: modelos de sobrecarga de presión en el VD en roedores muestran una reexpresión de genes fetales76,77 y alteraciones en proteínasmetabólicas, proteínas asociadas al estrés yestructurales78,79, aunque hay pocos modelos en animales grandes y no siempre concuerdan con los hallazgos en roedores80. Los datos respecto a humanos, obtenidos de biopsias endocárdicaspercutáneas, deberían interpretarse con precaución,dado que las alteraciones en el tabique del VD, delque suelen obtenerse las biopsias, pueden diferir delas alteraciones en la pared libre del VD, donde seproduce gran parte de la alteración estructural.
RESPUESTA DEL VENTRÍCULO DERECHO A UN AUMENTO CRÓNICO DEL VOLUMEN
La sobrecarga del volumen del VD debida a cortocircuitos intracardiacos o regurgitación tricuspídea o pulmonar suele tolerarse bien, tal vezporque el VD está adaptado para acomodargrandes cambios de volumen. Experimentalmente,la sobrecarga crónica del volumen del VD no parece afectar a la función contráctil del VD81, y clínicamente los pacientes con sobrecarga de volumendebida a cardiopatías congénitas parecen mantenerse bien durante muchos años82.
Sin embargo, la dilatación severa del VD impideen último caso un mayor aumento del volumen(bien debido a constricción pericárdica, bien acausa de la arquitectura compartida del VD y el VI), de forma que los incrementos adicionales en elvolumen del VD causan incrementos mínimos en lasuperficie de la pared libre del VD; en cambio, amedida que crece la presión del VD, se produce unaumento del volumen del VD a costa del VI. Estose ve con más claridad en la embolia pulmonar, enla que el aporte de volumen puede afectar negativamente a la función del VI83.
DESARROLLO DE INSUFICIENCIA CARDIACA DERECHA
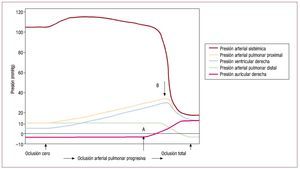
La insuficiencia cardiaca derecha, definida comola incapacidad del VD para generar un flujo continuo adecuado con una presión venosa centralnormal46, puede aparecer y ser grave en respuesta aaumentos en la resistencia vascular pulmonar pormuchas razones, como la obstrucción mecánica dela vasculatura pulmonar por embolia pulmonar opor vasoconstricción reactiva en respuesta a hipoxemia. Los datos experimentales obtenidos hacemás de cincuenta años mostraban que el VD tieneuna capacidad muy limitada para compensar talescargas. La figura 5 muestra el resultado de la oclusión progresiva de la arteria pulmonar en perroscon el tórax abierto84. Inicialmente, el aumento depresión en la arteria pulmonar se tolera bien, conuna presión sistémica prácticamente inalterada(probablemente debido al efecto Anrep, como yahemos analizado). En el punto A la presión venosacentral empieza a elevarse, lo que resulta en un incremento de contractilidad según la ley de Frank-Starling; cuando la presión del VD llega al nivel crítico en el punto B, la presión sistémica disminuyede manera repentina y catastrófica.

Fig. 5.Resultado de la oclusión progresiva de la arteria pulmonar de un perro con el tórax abierto. Inicialmente, un aumento progresivo en la presión dela arteria pulmonar se tolera bien, con una presión sistémica prácticamente inalterada. En el punto A la presión venosa central empieza a elevarse, loque permite la implicación de la función según la ley de Frank-Starling, hasta que la presión del VD alanza un nivel crítico en el punto B, donde la presiónsistémica decae de manera repentina y catastrófica. (Tomada de Guyton AC, et al. Circ Res. 1954; 2:326-32. Copyright 1954, con permiso de WoltersKluwer Health84). VD: ventrículo derecho.
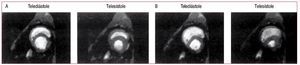
El mecanismo de insuficiencia del VD y colapsohemodinámico en este punto se debe a la interacción de varios factores. Justo antes del colapso hemodinámico, puede producirse una reducción delgasto cardiaco como consecuencia de la interaccióninterventricular. A medida que el VD se dilata enrespuesta a la sobrecarga de presión, la restriccióndel pericardio y de fascículos de fibras muscularescompartidas del VD y el VI limitan una mayor dilatación del VD y aumenta la pendiente de la curvade relación presión-volumen diastólica del VD, demodo que a un mayor incremento en la presión delVD le corresponde un menor estiramiento de lapared libre del VD y, por lo tanto, menos reclutamiento de la función según la ley de Frank-Starling.Al mismo tiempo, el desplazamiento del septo interventricular disminuye la eyección del VI. La combinación deriva en una reducción neta del gasto cardiaco. La figura 6 muestra vistas del eje corto decorazones de perro sometidos a embolias pulmonares experimentales: gran parte del aumento delvolumen del VD se produce a costa del volumen delVI85.

Fig. 6. Imágenes del eje corto del ventrículo de un perro obtenidas utilizando imágenes de resonancias magnéticas en diástole y en sístole, en condicionesprevias al estudio (A) y tras la embolización pulmonar experimental (B). El aumento en la sobrecarga de presión del VD conduce a una dilatación diastólicadel VD y un desplazamiento septal hacia el VI, lo que pone en peligro el llenado del VI y reduce el gasto del VI. (Tomada de Dell'Italia LJ. J Appl Physiol.1995;78:2320-7. Copyright 1995, con permiso de la Sociedad Americana de Fisiología85). VD: ventrículo derecho; VI: ventrículo izquierdo.
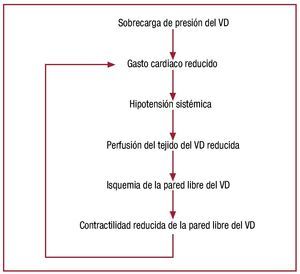
Una vez que el gasto cardiaco comienza a declinar, el colapso hemodinámico avanza rápidamente. La figura 746 esquematiza el mecanismo másprobable de colapso hemodinámico: el aumento dela presión del RV debido a un aumento de la poscarga deriva en una reducción del gasto cardiaco ehipotensión sistémica; ello reduce la presión de perfusión del tejido del VD, que cuando desciende aniveles críticos, produce isquemia de la pared libre del VD86,87. La isquemia del VD produce una reducción de la función contráctil, lo que afecta a la capacidad del VD para manejar la poscarga elevadadel VD, reduce el gasto cardiaco e inicia una rápidaespiral descendente que progresa hacia el colapsohemodinámico.

Fig. 7. Esquema propuesto para el mecanismo de descompensación hemodinámica repentina en el contexto de un aumento grave de la impedancia pulmonar de entrada. Una vez que la resistencia alcanza el nivel B enla figura 5, la salida del VD desciende a niveles críticos, lo que conduce auna hipotensión sistémica, precipitación de isquemia del VD, declive de lafunción contráctil intrínseca del VD, descenso de la salida del VD y una espiral descendente que progresa hasta el colapso hemodinámico y la muerte. Sin una intervención para reducir la impedancia pulmonar o aumentarel gasto cardiaco, este proceso sería irreversible. (Tomada de Greyson CR.Crit Care Med. 2008;36:S57-65. Copyright 2008, con permiso de Lippincott,Williams & Wilkins46). VD: ventrículo derecho.
Dado que el colapso hemodinámico en este escenario suele ser repentino (y con frecuencia irreversible) y que la insuficiencia cardiaca derecha no semanifiesta en una presión venosa central elevadahasta que la compensación del VD está casi agotada, muchos (si no la mayoría) de los pacientes quepresentan síntomas y signos de insuficiencia cardiaca derecha pueden hallarse en una situación hemodinámica límite, en la que la presión venosa central ha comenzado a elevarse pero la presiónsistémica aún no ha comenzado a descender (entrelos puntos A y B de la fig. 5). Evidencias experimentales recientes indican que la disfunción contráctil progresiva del VD no relacionada con la isquemia desemboca en sobrecarga aguda de presiónen el VD25,88-90 cercana a este límite y puede contribuir al repentino deterioro hemodinámico en pacientes con aparente estabilidad hemodinámica.
Hay varios mecanismos que pueden contribuir ala disfunción progresiva del VD en este contexto.Las anomalías del acoplamiento ventriculovascularpueden reducir la eficiencia de la transmisión deenergía del VD a la circulación pulmonar25. Mientras que algunos investigadores creen que laisquemia crónica desempeña un papel en la insuficiencia cardiaca derecha, las intervenciones paraaumentar la perfusión coronaria del VD en modelos de insuficiencia inducida por sobrecarga depresión no han mejorado de forma significativa lafunción contráctil del VD91 y es evidente que la disfunción contráctil progresiva puede producirse enausencia de signos de isquemia25,88-90. La activación de proteasas intracelulares como la calpaína92 o la activación de vías apoptóticas (muerte celularprogramada)93,94 pueden contribuir a la disfuncióndel VD. La inhibición de la calpaína parece atenuar la disfunción92 y reducir la apoptosis del VD94 asociada a sobrecargas de presión. La calpaína puedeproducir disfunción contráctil a través de la degradación de proteínas de adhesión intercelular comola talina, que coordina la contracción cardiaca95,96.
Un trabajo reciente ha identificado la tenascina,una proteína de unión a la matriz extracelular,como posible causa de insuficiencia cardiaca crónica97. Se desconoce si los cambios mencionados enla expresión génica son adaptativos o maladaptativos.
IMPLICACIONES PARA LA INVESTIGACIÓN FUTURA
En teoría, la progresiva disfunción contráctil podría alterar el umbral en el que comienza la espiraldescendente al colapso hemodinámico. Tal vez estopodría explicar por qué los pacientes con situaciones hemodinámicas pulmonares aparentementeestables pueden descompensarse repentinamente.Como posibilidad alternativa, la disfunción contráctil del VD podría hacer que un paciente fueramás susceptible al colapso hemodinámico tras unevento posterior que comprometiera aún más lacontractilidad del VD. Un mejor conocimiento delmecanismo de la insuficiencia del VD en el contextode la HP podría acrecentar el interés en este trastorno actualmente desatendido, y podría facilitar eldesarrollo de nuevos enfoques terapéuticos quevayan más allá de la simple reducción de la impedancia vascular pulmonar y empiecen a abordar losmecanismos subyacentes a la insuficiencia del VD.
ABREVIATURAS
Ea: elastancia arterial efectiva.
Emáx: elastancia máxima.
Esf: elastancia telesistólica.
HP: hipertensión pulmonar.
RVP: resistencia vascular pulmonar.
VD: ventrículo derecho.
VI: ventrículo izquierdo.
Full English text available from: www.revespcardiol.org
Financiado por el United States Department of Veterans Affairs, y elUnited States National Heart, Lung and Blood institute, becas R01-HL068606 and R01-HL49444
Correspondencia: Dr. C.R. Greyson.
Cardiology 111B, Denver VAMC.
1055 Clermont Street. Denver, CO 80220. Estados Unidos.
Correo electrónico: Clifford.Greyson@UCDenver.edu; Clifford.Greyson@VA.gov