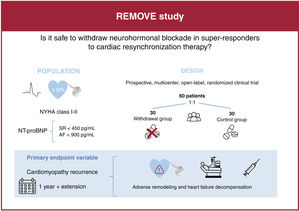

Cardiac resynchronization therapy (CRT) is an effective treatment for patients with nonischemic dilated cardiomyopathy associated with left bundle branch block (LBBB). In these patients, the device can normalize left ventricular ejection fraction (LVEF). Nevertheless, it remains unclear whether CRT responders still require neurohormonal blockers. The aim of this study is to determine the long-term safety of withdrawing drug therapy in these patients.
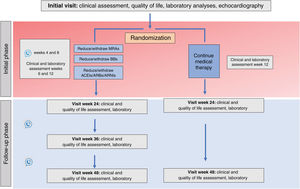
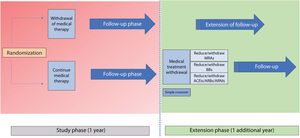
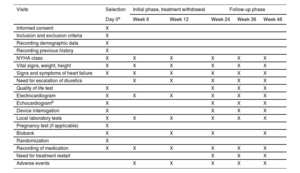
MethodsThe REMOVE trial is a prospective, multicenter, open-label and randomized 1:1 study designed to assess the effect of withdrawing neurohormonal blockers in patients with nonischemic dilated cardiomyopathy associated with left bundle branch block who recovered LVEF after CRT. The study will include a 12-month follow-up with the option to continue into the follow-up extension phase for up to 24 months. The primary endpoint is the recurrence of cardiomyopathy defined as any of the following criteria: a) a reduction in LVEF >10% (provided the LVEF is <50%); b) a reduction in LVEF >10% accompanied by an increase >15% in the indexed end-systolic volume relative to the previous value and in a range higher than the normal values, or c) decompensated heart failure requiring intravenous diuretic administration. In patients meeting the primary endpoint, drug therapy will be restarted.
ConclusionsThe results of this study will help to enhance our understanding of CRT superresponders, a specific group of patients.
Registred at ClinicalTrials.gov (Identifier: NCT05151861).
Keywords
Identify yourself
Not yet a subscriber to the journal?
")
Purchase access to the article
By purchasing the article, the PDF of the same can be downloaded
Price: 19,34 €
Phone for incidents
Monday to Friday from 9am to 6pm (GMT+1) except for the months of July and August, which will be from 9am to 3pm