Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a heritable disease characterized by the appearance of polymorphic ventricular tachycardia during exercise, emotion, or catecholamine perfusion.1 This cardiopathy is considered a rare disease, with a prevalence of 1/10 000, and is a highly lethal entity (30% of sudden deaths in individuals under 40 years of age who are not undergoing treatment with beta-blockers2). It is usually characterized by autosomal dominant inheritance with 80% penetrance, and by mutations in the ryanodine receptor 2 gene (RYR2).3 Diagnosis usually requires an exercise stress test (EST) or adrenaline test.4–6
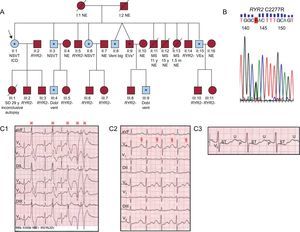
Our objective is to describe a kindred with 19 living members (Figure A), 4 with sudden death and 8 carriers of the new RYR2 C2277R variant (genotype+) (Figure B), 7 of whom exhibit the CPVT phenotype according to EST results (phenotype+).
 of the first minute of recovery following the exercise stress test in the proband, showing polymorphic ventricular arrhythmias (C1, ) and notable merging of the U-wave with the subsequent P wave (C2, §). CD, implantable cardioverter defibrillator; Dobl vent, ventricular doublets; m, month; NE, not evaluated; NSVT, nonsustained ventricular tachycardia; SD, sudden death; Vent big, ventricular bigeminy; Dobl vent, ventricular doublets; VEs, ventricular extrasystoles; y, years.")
A: Family tree; the arrow indicates the proband; the blue symbols represent patients with the catecholaminergic polymorphic ventricular tachycardia phenotype, and asterisks indicate heterozygous carriers of the RYR2 C2277R mutation. B: Electropherogram of the fragment of exon 45 of the RYR2 gene containing the mutation. C: Consecutive records (C1-C3) of the first minute of recovery following the exercise stress test in the proband, showing polymorphic ventricular arrhythmias (C1,
The proband (II: 1), aged 56, presented with syncope and palpitations. She reported the sudden death of 3 siblings, aged 11 and 15 years (due to physical exercise and an argument) and 1.5 months, and a daughter aged 29 years (while dancing, with previous syncope during exertion), although autopsy was only performed in the latter case, and was inconclusive. We applied the protocol approved by our ethics committee for family studies following sudden death in individuals with unknown cause, and all individuals gave informed consent. In the proband, the electrocardiogram and echocardiogram were normal. Her EST (Bruce protocol) showed ventricular arrhythmias at 100 bpm and above, and was diagnostic of CPVT (Figure C). This diagnosis was defined by the presence of ventricular doublets, sustained ventricular tachycardia, or non-sustained polymorphic ventricular tachycardia or > 10 premature ventricular contractions/min during the EST or adrenaline test.6 Six other members of the 19 tested showed a similar response, with different degrees of complexity of the ventricular arrhythmias (Figure A, Table). Suspecting autosomal dominant CPVT, we sequenced the 33 most frequently affected exons of the RYR2 gene in the proband. We identified a previously unreported heterozygous missense variant in exon 45 (C2277R), located in a hot spot encoding part of the calstabin-binding domain, which was classified as a mutation that is probably associated with the disease. The mutation cosegregated with the CPVT phenotype, although in 1 person (II: 9), who also underwent an adrenaline test (Mayo Clinic protocol), ventricular arrhythmias did not meet the diagnostic criteria. Thus, we obtained a cohort of 8 carrier subjects, 7 of whom were found by EST to have the CPVT phenotype (87.5% penetrance). Of the carriers, 75% were male, mean age 46 years (SD, 10), 37% reported prior arrhythmic symptoms, and 100% had a normal electrocardiogram at rest (heart rate, 63 [SD,10] bpm, QTc 400 [SD,27] ms). In the initial EST (duration 9 [SD,2] min), the maximum heart rate was 150 [SD,15] bpm, and the diagnosis was established at a heart rate of 132 (SD,11) bpm.
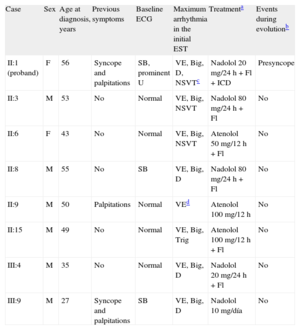
Clinical Characteristics of the Cohort of Eight Family Members Carrying the Mutation in RYR2
| Case | Sex | Age at diagnosis, years | Previous symptoms | Baseline ECG | Maximum arrhythmia in the initial EST | Treatmenta | Events during evolutionb |
| II:1 (proband) | F | 56 | Syncope and palpitations | SB, prominent U | VE, Big, D, NSVTc | Nadolol 20 mg/24 h + Fl + ICD | Presyncope |
| II:3 | M | 53 | No | Normal | VE, Big, NSVT | Nadolol 80 mg/24 h + Fl | No |
| II:6 | F | 43 | No | Normal | VE, Big, NSVT | Atenolol 50 mg/12 h + Fl | No |
| II:8 | M | 55 | No | SB | VE, Big, D | Nadolol 80 mg/24 h + Fl | No |
| II:9 | M | 50 | Palpitations | Normal | VEd | Atenolol 100 mg/12 h | No |
| II:15 | M | 49 | No | Normal | VE, Big, Trig | Atenolol 100 mg/12 h + Fl | No |
| III:4 | M | 35 | No | Normal | VE, Big, D | Nadolol 20 mg/24 h + Fl | No |
| III:9 | M | 27 | Syncope and palpitations | SB | VE, Big, D | Nadolol 10 mg/día | No |
Big, bigeminy; D, doublets; ECG, electrocardiogram; EST, exercise stress test; F, female; Fl, flecainide; ICD, implantable cardioverter defibrillator; M, male; NSVT, nonsustained ventricular tachycardia with 180° change of axis; SB, sinus bradycardia; Trig, trigeminy; VE, ventricular extrasystoles.
The carriers were treated with beta-blockers at the maximum tolerated dose, and while the ventricular arrhythmias disappeared during the follow-up EST in 3 subjects (37%), in the remaining 5 (63%) the arrhythmic burden (frequent ventricular premature beats, bigeminy, doublets, nonsustained ventricular tachycardia) persisted enough to add flecainide to the treatment regime, as previously proposed1 (Table). The proband was implanted with a defibrillator due to presyncope with nonsustained ventricular tachycardia during the EST, despite maximum treatment with beta-blockers (before starting the use of the flecainide treatment in this clinical context1). Finally, at 34 (SD,4) months follow-up, all patients were asymptomatic, without arrhythmia or remarkable clinical events (sudden death, syncope, or appropriate defibrillator discharge).
In summary, for the first time we describe the RYR2 C2277R mutation as a cause of CPVT, in a family with high lethality in younger individuals, with a good diagnostic yield using EST and an excellent response to treatment with beta-blockers, with and without flecainide.
FUNDINGThis work was funded by the Instituto de Salud Carlos III (PI14/01477 y RD12/0042/0029), Prometeo 2011/027, the Sociedad Española de Cardiología (Pedro Zarco Scholarship) and the Agence Nationale de la Recherche (ANR-13-BSV1-0023-03).
We are grateful for the kind cooperation of the patients, and the working group on sudden infantile death of Spanish Association of Pediatrics, and for technical support from the La Fe Biobank (PT13/0010/0026).